

Para evitar confusiones, le informamos de que ESTE NO ES el formulario para enviar PREGUNTAS SOBRE VACUNAS al comité. Si esa fuera su intención, diríjase a la sección de preguntas de profesionales o de preguntas de familias.
A través de este formulario, el responsable del mismo, la Asociación Española de Pediatría (AEP) y su Comité Asesor de Vacunas, recaba los datos necesarios para gestionar el envío de sus comentarios y sugerencias sobre el Manual de Inmunizaciones en línea de la AEP, que ponemos a su disposición en nuestra página web.
Los campos marcados con asterisco son obligatorios y, sin ellos, no se tramitará su comentario o sugerencia.
Este tratamiento de datos no puede realizarse sin su consentimiento, por lo que deberá validar la casilla de protección de datos antes de enviar la consulta.
Sus datos no serán cedidos a otras entidades, ni transmitidos a otros países. Tiene derecho a acceder, rectificar y suprimir los datos, así como otros derechos, como se explica en la información común a los tratamientos que efectúa la AEP.
Puede consultar la información detallada sobre protección de datos, así como la información común a los tratamientos que efectúa la AEP.
47. Historia de las inmunizaciones
CAPÍTULO 47 - HISTORIA DE LAS INMUNIZACIONES
2.1. Real Expedición Filantrópica de la Vacuna, 1803-1808
3.1. Las vacunas atenuadas contra el cólera aviar
3.2. Primera vacuna en seres humanos: la vacuna contra la rabia
- BCG: primera vacuna contra la tuberculosis
- De la atenuación del patógeno a las vacunas de subunidades
5.1. Desarrollo de la vacuna contra la fiebre amarilla 17D
5.2. Antitoxinas y toxinas: los toxoides como vacunas
5.3. Primeras vacunas combinadas: DTP (difteria, tétanos y tosferina)
5.4. Vacuna de la poliomielitis: de la vacuna inactivada de Salk a la atenuada de Sabin
5.5. Otras vacunas víricas: vacuna triple vírica. Sarampión, rubeola, paperas (SRP)
- Vacunas producidas por ADN recombinante: hepatitis B
- Vacunas conjugadas de polisacáridos
- La vacunología reversa
- Vacunas en el siglo XXI
9.1. Logros y retos
9.2. Vacunas contra la COVID-19. Vacunas ARNm, los vectores virales se hacen realidad
9.3. Vacunación frente al VRS. La inmunización con anticuerpos, de la sueroterapia a los anticuerpos monoclonales
9.4. Retos futuros en vacunas
- Bibliografía
- Enlaces de interés
- Historial de actualizaciones
- Figuras incluidas en el capítulo:
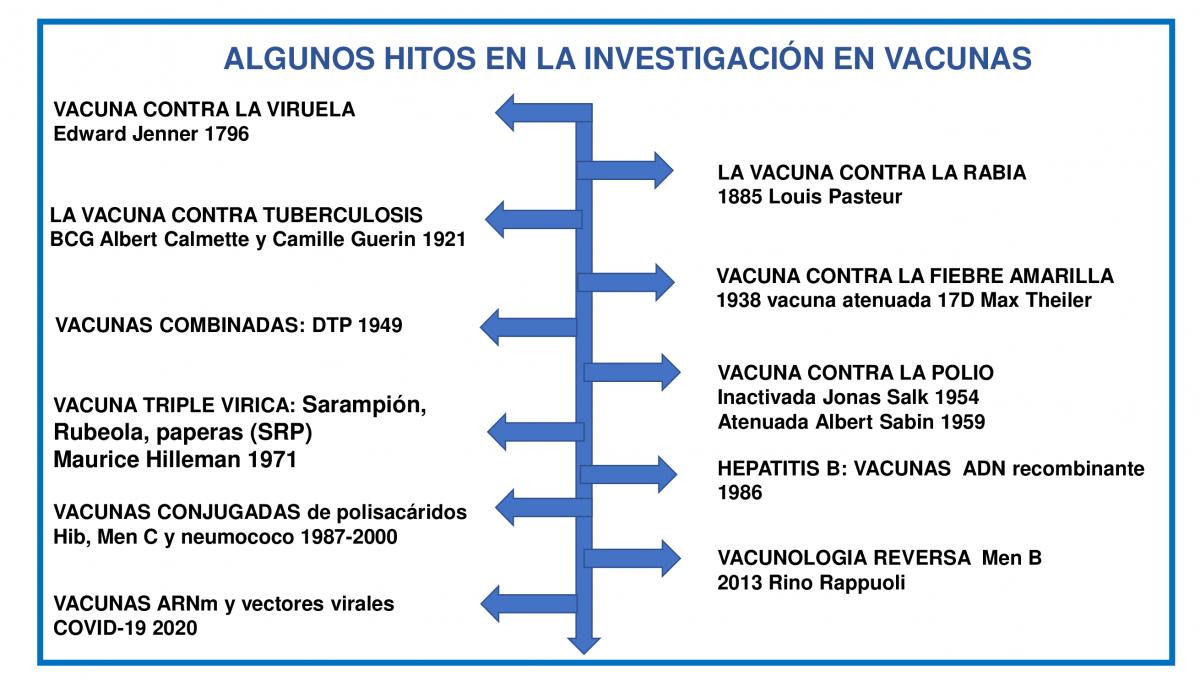
Figura 47.1. Hitos en la investigación en vacunas
Figura 47.2. La primera vacunación contra la viruela Edward Jenner, 1796
Figura 47.3. Real Expedición Filantrópica de la Vacuna, 1803-1808
Figura 47.4. Jenner descubre la vacunación y Pasteur las vacunas
Figura 47.5. BCG la primera vacuna contra la tuberculosis con 100 años de historia
Figura 47.6. Plataformas para el desarrollo de nuevas vacunas
Sugerencia para la citación: Comité Asesor de Vacunas e Inmunizaciones (CAV-AEP). Historia de las inmunizaciones. Manual de inmunizaciones en línea de la AEP [Internet]. Madrid: AEP; feb/2026. [consultado el dd/mmm/aaaa]. Disponible en: http://vacunasaep.org/documentos/manual/cap-47
1. Introducción.
En el año 1900, la esperanza de vida al nacer en España apenas alcanzaba los 35 años. En 2024, la esperanza de vida media en España era de 84,01 años y en la Unión Europea-27, de 81,7 años. En América Latina y Caribe este indicador ha presentado un incremento similar, pasando de 29 años en 1900 a los 76 años actuales. Esta mejora de la esperanza de vida y de la salud poblacional responde a un origen multifactorial, en el que confluyen el desarrollo económico, las mejoras en las condiciones higiénico-sanitarias, el descubrimiento y uso de los antibióticos, así como la implementación de programas generalizados de vacunación.
Los avances conseguidos en el control de numerosas enfermedades infecciosas y la historia del descubrimiento y el impacto las vacunas, resulta apasionante. El primer avance científico en este campo se produjo cuando Edward Jenner, en 1796, utilizó un virus animal denominado viruela vacuna (Cowpox virus) para proteger de la enfermedad causada por el virus humano (Variola virus). Louis Pasteur, casi 100 años más tarde, logró atenuar patógenos humanos permitiendo su utilización como vacunas. Hoy disponemos de vacunas para prevenir más de 20 enfermedades que pueden ser mortales.
Clásicamente, desde el descubrimiento de una vacuna, hasta su incorporación a la práctica clínica transcurrían una media de 15 a 20 años. El virus ébola y, más recientemente, la alerta sanitaria que produjo el virus SARS-CoV-2, han modificado sensiblemente este hecho. Las mejoras tecnológicas y regulatorias, mostraron que resulta factible acelerar el descubrimiento y la puesta en uso de nuevas vacunas, manteniendo los más elevados estándares de calidad y seguridad.
Es de destacar que la aparición en los últimos años de microorganismos multirresistentes a los antimicrobianos ha venido a incrementar el valor añadido de las vacunas. Es importante, por lo tanto, comprender el recorrido histórico de las inmunizaciones, no solo para aportar perspectiva crítica, sino para facilitar reconocer patrones de innovación, anticipar desafíos y diseñar estrategias más eficaces, equitativas y sostenibles para el desarrollo de las futuras generaciones de las inmunizaciones.
Figura 47.1. Hitos en la investigación en vacunas.
2. PRIMERA VACUNA: VIRUELA (eng: smallpox; fr: variole)
La viruela humana es una enfermedad muy contagiosa causada por el Orthopoxvirus variola. A la familia Orthopoxvirus pertenecen, entre otros, los virus de la viruela bovina, humana y el virus de la viruela del mono (monkeypox).
Se han encontrado numerosas pruebas que reflejan que desde el año 1000 de nuestra era, en China, África y Turquía, se realizaba la variolización. Este proceso consistía en inocular material extraído de las pústulas de la viruela humana en personas sanas. Jenner, utilizando la observación y aplicando el método científico, demostró que la exposición a una forma menor de la enfermedad, la viruela bovina de las vacas (Cowpox virus), protegía de la forma grave y mortal de la enfermedad, la viruela de humanos (Variola virus). Jenner comprobó además que la linfa vacunal de la viruela bovina podía transferirse entre individuos conservando su capacidad protectora. En 1798 documentó que las personas vacunadas -como el niño Phipps- no enfermaban tras la exposición posterior a la viruela humana.
Figura 47.2. La primera vacunación contra la viruela Edward Jenner, 1796.

Modificada del libro de Jenner “Vaccination against smallpox”. 1796.
2.1. Real Expedición Filantrópica de la Vacuna, 1803-1808
Para erradicar una enfermedad se precisa no sólo el descubrimiento de una vacuna sino su “distribución universal”. En noviembre de 1803 partió del puerto de La Coruña la “Real Expedición Filantrópica de la Vacuna”, promovida por el rey Carlos IV, con el objetivo de introducir la vacuna en América y Asia. El itinerario del buque “María Pita” fue desde la Coruña a Canarias, Venezuela, Colombia, Ecuador, Perú, México, Filipinas y China, siendo esta, la primera expedición humanitaria global de la historia.
La misión estaba dirigida por los médicos Francisco J. Balmis y José Salvany, quienes idearon una ingeniosa forma para transportar y conservar la vacuna, el brazo de los 22 niños que participaron en la expedición. La vacuna se inoculaba, de brazo en brazo, durante la travesía del Atlántico. Estos niños estaban al cuidado de Isabel Zendal, directora del orfanato de Coruña y madre de uno de ellos. Isabel Zendal ha sido considerada, por la Organización Mundial de la Salud (OMS), como la primera enfermera en misión humanitaria.
Figura 47.3. Real Expedición Filantrópica de la Vacuna, 1803-1808.

Modificada de Soto-Pérez-de-Celis E. Postgrad Med J. 2008.
Se estima que esta expedición permitió la vacunación de más de medio millón de personas. Jenner escribió “No puedo imaginar que en los anales de la Historia se proporcione un ejemplo de filantropía más noble y más amplio que está expedición de la vacuna”. La historia de esta expedición, de enorme importancia para la salud pública, es, aún hoy, escasamente conocida y poco reconocida.
La OMS declaró oficialmente erradicada la viruela en 1980. Hasta la fecha, continúa siendo la única enfermedad infecciosa humana erradicada a nivel mundial gracias a la vacunación, si bien determinados serotipos de poliovirus, como los tipos 2 y 3, también han sido erradicados.
3. LAS VACUNAS DE JENNER A PASTEUR
3.1. Las vacunas atenuadas contra el cólera aviar (eng: avian cholera, fr: cholera aviaire) y carbunco (eng: anthrax; fr: antharx)
El químico Louis Pasteur inició sus trabajos en vacunas casi 100 años después de los experimentos de Jenner. A él debemos la denominación de vacuna, como guiño a su herencia bovina y a su origen (vacca), e importantes hallazgos que lo han convertido en el “inventor” de las vacunas.
En sus inicios sus trabajos se orientaron hacia las enfermedades infecciosas que afectaban a animales. En 1879 hizo su primer descubrimiento aislando la Pasteurella multocida, el agente causal del cólera aviar. Observó que, un cultivo antiguo y olvidado en el laboratorio, había perdido su virulencia y conseguía proteger a las gallinas del cólera aviar. Pasteur llamó a esta pérdida progresiva de virulencia "atenuación". Este término se continúa utilizando, actualmente, para denominar a las vacunas que utilizan una variante viva atenuada del patógeno.
En 1881, Pasteur comenzó a trabajar en la vacuna contra el carbunco, enfermedad del ganado que se transmitía por las esporas de la bacteria Bacillus anthracis. Descubrió que los cultivos de ántrax crecían fácilmente, a una temperatura de 42 a 43 °C, perdiendo su capacidad para formar esporas. Observó que estos cultivos no esporulantes podían mantenerse a esa temperatura, durante 4-6 semanas, y que, si se inoculaban en animales, mostraban una marcada disminución de la virulencia. Pasteur consiguió, así, la atenuación de B. anthracis y demostró que los carneros vacunados con cultivos atenuados y, posteriormente inoculados con el patógeno virulento, sobrevivían mientras que los no vacunados morían. Este experimento representó un auténtico reto científico en la época.
Figura 47.4. Jenner descubre la vacunación y Pasteur las vacunas.

Modificado de Nature Reviews Microbiology 2007. Stefan H.E. Kaufmann.
3.2. Primera vacuna en seres humanos: la vacuna contra la rabia (eng: rabies; fr: rage)
La rabia es la zoonosis viral conocida más antigua. Es una enfermedad aguda infecciosa ocasionada por un Rhabdoviridae que se localiza en la saliva y secreciones de los animales infectados. Sus principales huéspedes y transmisores son los perros. Las personas se infectan por la mordedura o el arañazo de un animal infectado. Ocasiona un cuadro de encefalitis aguda y presenta una letalidad cercana al 100 %.
Pasteur había realizado sus experimentos de atenuación del virus. Inyectar a un humano un agente patógeno, aunque debilitado, fue objeto de una importante controversia por no haberse realizado previamente. Pasteur descubrió que la virulencia de las cepas del virus de la rabia disminuía, cuando el material infectado se inyectaba en diferentes especies animales. Partiendo de una cepa altamente virulenta, Pasteur consiguió, por sucesivos pases en médula espinal de conejo, atenuar el virus de la rabia y, su posterior utilización como vacuna terapéutica en humanos. En 1885, en su informe a la Academia de Ciencias de Francia, detalló todos estos pasos. Un año más tarde presentó un documento con los resultados de la inoculación de su vacuna en 350 personas. Se registró una muerte y consideró que, la vacunación tardía no permitió prevenir la enfermedad.
El éxito de estas vacunaciones convirtió a Pasteur en un héroe nacional, facilitando la creación del Instituto Vacunal contra la Rabia, inaugurado en París en 1888, como Dispensario contra la Rabia, Centro de Investigación de Enfermedades Infecciosas y Centro Docente Superior. Hoy el “Instituto Pasteur”, es centro de referencia en investigación biomédica y cuenta con una red de 30 Institutos Pasteur distribuidos por 27 países de los cinco continentes.
4. BCG: LA PRIMERA VACUNA CONTRA LA TUBERCULOSIS (eng: tuberculosis, fr: tuberculose)
Los últimos años del siglo XIX constituyeron una etapa emocionante para el progreso de la medicina. No sólo Shibasaburō Kitasato y Emil von Behring habían demostrado las propiedades antimicrobianas del suero de los convalecientes; Pasteur había conseguido una serie de vacunas atenuadas; y, en 1882, Robert Koch había identificado Mycobacterium tuberculosis como el agente infeccioso causal de la tuberculosis.
En este contexto, en 1900 en el Instituto Pasteur de Lille, el médico Albert Calmette y el veterinario Camille Guérin iniciaron una estrecha colaboración buscando una vacuna contra la tuberculosis. Sus esfuerzos iniciales se centraron en cultivar una cepa patógena de Mycobacterium bovis, agente causal de la tuberculosis bovina, y atenuarla en el laboratorio (in vitro). Trece años más tarde, y después de 230 pases de subcultivos, lograron una cepa viva y atenuada y, que no causaba enfermedad probada en una amplia variedad de animales incluidos cobayas, monos, terneros y caballos. Esta cepa, genéticamente muy distante de su ancestro patógeno, fue denominada Bacille Calmette-Guérin (BCG).
Figura 47.5. BCG la primera vacuna contra la tuberculosis con 100 años de historia.
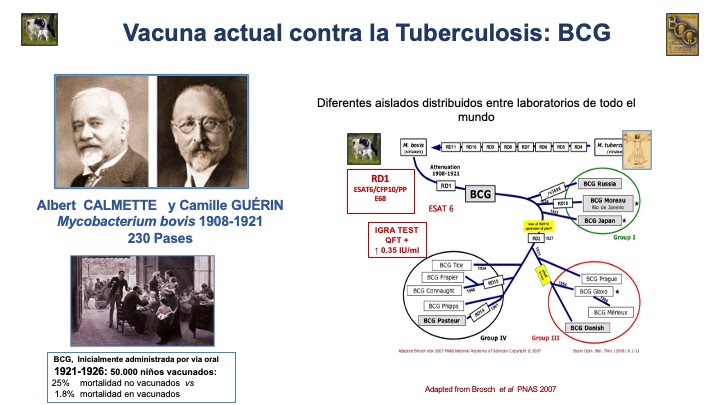
Modificado de Colección Instituto Pasteur y PNAS. Brosch, et al . 2007
La secuenciación genética ha mostrado que la atenuación de BCG se debe a una pérdida en el genoma de la bacteria, concretamente, en la región denominada RD1 (Región Diferencial 1). En esta región se localiza el gen que codifica para ESAT-6 que, cuando se secreta, es uno de los mayores factores de virulencia del complejo M. tuberculosis, pero junto a otras dos proteínas codificadas en esta delección, CFP10 y PPE 68, son unos potentes antígenos que están conservados en las cepas del complejo y su pérdida puede ser una de las causas de la falta de protección de BCG contra la tuberculosis pulmonar.
En 1921 se vacunó con BCG, por vía oral, al primer bebé cuya madre había muerto de tuberculosis poco después de su nacimiento. En los seis años siguientes se vacunaron más de 50 000 lactantes demostrando que la vacuna viva atenuada BCG era segura y que podría proteger frente a otras enfermedades infecciosas. Este efecto “no específico” de las vacunas vivas atenuadas se ha asociado a una reducción global de la mortalidad infantil, atribuida no solo a la prevención de la tuberculosis, sino también a una protección frente a otras infecciones, con tasas de mortalidad descritas en torno al 25 % en no vacunados frente a menos del 2 % en vacunados.
A finales de la década de 1920 la cepa vacunal BCG se distribuyó por todo el planeta. Los laboratorios mantuvieron la vacuna mediante pases sucesivos en medios de cultivo. Estos pases fueron el origen de los cambios observados entre las diferentes subcepas BCG designadas con el nombre del laboratorio (BCG Pasteur) o geográfica (BCG Russia, Danish y Japan). En los últimos años, se han realizado estudios comparativos de estas subcepas BCG, mostrando diferencias en la morfología y en la respuesta inmunitaria mediada por células. Es por este motivo que la OMS recomienda el mantenimiento de BCG en forma liofilizada para la producción de vacunas, con el fin de preservar su estabilidad genética e inmunogenicidad.
Hoy, 100 años después, BCG es la vacuna más administrada en el mundo y forma parte de la lista de medicamentos esenciales de la OMS. Sin embargo, su aceptación y recomendación es irregular entre continentes, con menor utilización en el mundo industrializado. Entre las principales razones que explican estas diferencias se encuentran el menor riesgo de transmisión de la tuberculosis y el acceso a los servicios esenciales de atención sanitaria y a su tratamiento. Sin duda, también contribuye a su no-utilización la controversia existente en torno a la efectividad de la BCG. Esta vacuna se ha mostrado útil contra la tuberculosis diseminada y la meningitis tuberculosa en la infancia y relativamente pobre contra la forma más común de la enfermedad en adultos, la tuberculosis pulmonar. Los estudios publicados y algún metaanálisis reciente indican una protección muy variable frente a esta forma responsable de la transmisión respiratoria, con una efectividad del 80 % en nativos americanos y niños británicos y una escasísima protección en los estudios de Madrás, India.
Por otra parte, BCG parece tener efectos beneficiosos indirectos no específicos, a través de la estimulación generalizada del sistema inmunitario, que puede proteger contra otros patógenos. Desde la década de 1960, BCG ha demostrado su valor como inmunomodulador, específicamente en la quimioterapia de los cánceres de vejiga. En los últimos años se ha consolidado la evidencia sobre los efectos heterólogos de la vacuna BCG, demostrando que induce inmunidad entrenada mediante reprogramación epigenética y metabólica de células del sistema inmune innato, potenciando la respuesta frente a patógenos no relacionados y reduciendo la morbimortalidad por infecciones diversas. Estos hallazgos han abierto nuevas perspectivas en la inmunomodulación y en el desarrollo vacunal.
Actualmente y, a pesar de que existe un tratamiento antibiótico efectivo, la tuberculosis continúa siendo la enfermedad infecciosa que causa más muertes en el mundo. Desde los años 90, la asociación de VIH/SIDA y tuberculosis y la aparición de cepas resistentes han convertido en una prioridad conseguir una nueva vacuna que proteja contra las formas respiratorias. En el año 2023, 14 nuevas vacunas se encuentran en distintas fases de ensayos clínicos, con el objetivo de proteger contra las formas respiratorias de la enfermedad y disminuir su transmisión, siendo la vacuna española MTBVAC, basada en el patógeno atenuado de origen humano, una de las más avanzadas en estudios clínicos.
5. DE LA ATENUACIÓN DEL PATÓGENO A LAS VACUNAS DE SUBUNIDADES
5.1. Desarrollo de la vacuna contra la fiebre amarilla 17D. (eng: yellow fever; fr: fièvre jaune)
La fiebre amarilla (FA) es una enfermedad vírica, endémica en gran parte del África subsahariana y la región de las Américas, causada por un flavivirus y propagada por el mosquito Aedes aegypti.
En 1930, el virólogo sudafricano Max Theiler, comenzó sus trabajos buscando reducir la patogenicidad del virus, y su posible utilización como vacuna. Aisló el virus de la sangre de un ghanés llamado Asibi y constató que el suero de pacientes con fiebre amarilla protegía a los monos de la infección, aunque el virus muerto no inducía inmunidad. En 1937, Max Theiler y Hugh Smith, describieron el desarrollo de una cepa viva atenuada de vacuna contra la FA utilizando huevos de gallina embrionados. Partiendo de la cepa Asibi, aislaron la cepa 17D tras 176 pases, inicialmente, en tejido embrionario de ratón y suero de mono y, ya posteriormente, en embrión de pollo. Su inoculación en Macacus rhesus mostró que la vacuna producía anticuerpos a los 14 días e inmunidad a la infección en una semana. La cepa atenuada 17D había perdido el neurotropismo y conservaba el potencial de desencadenar una respuesta inmune.
La cepa atenuada 17D (YF17D) recibió, en 1938, la aprobación para su uso como vacuna. Se actualizó en 1970, siendo en gran medida, la que se utiliza para prevenir la enfermedad en población de riesgo. Se trata de una vacuna que genera una rápida y muy intensa respuesta inmunitaria adaptativa y con duración básicamente de por vida. Los eventos adversos graves, neurotrópicos y viscerotrópicos, son infrecuentes. En 1951, Max Theiler recibió el Premio Nobel de Fisiología o Medicina por su vacuna 17D por "sus descubrimientos sobre la fiebre amarilla y cómo combatirla”.
Evaluación de las respuestas a las vacunas: Aplicación de la Biología de Sistemas para comprender su mecanismo de acción
El desarrollo de la biología de sistemas permitió comprender, 70 años más tarde, el mecanismo de protección de esta vacuna viva atenuada. En 2008, se publicaron dos artículos fundamentales para la inmunología de vacunas, en los que evaluaban la respuesta del sistema inmunitario a la vacuna 17D (YF17D) y que convertían, a esta vacuna, en la candidata ideal para el estudio de las respuestas inmunitarias innatas y adaptativas a la vacunación. Bali Pulendran y cols, en su artículo publicado en Nature Immunology, identificaron las firmas inmunitarias innatas que, mediante una combinación de citometría de flujo de parámetros múltiples, análisis de citoquinas y de quimiocinas por Multiplex, podrían utilizarse para la predicción de respuestas inmunitarias adaptativas posteriores. Este enfoque múltiple les permitió identificar una firma genética que, con un 90 % de exactitud, predice la respuesta de células T CD8+ y una segunda firma que, con un 100 % de precisión, predice la respuesta de anticuerpos neutralizantes a la vacuna. Otros importantes hallazgos del equipo de Bali Pulendran, en la respuesta innata a YF17D son el complemento, el receptor tipo Toll 7 (TLR7) y la vía de señalización del interferón tipo I.
Posteriormente, la modelización de las respuestas de anticuerpos neutralizantes y de las células T citotóxicas, permitió identificar y obtener una visión global de la respuesta del sistema inmunitario a la vacunación. Por todo ello, el estudio de la inmunidad protectora de YF17D, se ha considerado como el inicio de la vacunología de sistemas como campo de investigación.
Este enfoque de biología de sistemas también se ha utilizado para predecir la respuesta inmunitaria a otras vacunas, como la vacuna contra la gripe estacional. Añadir que el importante volumen de datos generados por estos estudios ha alentado colaboraciones a gran escala y a la implementación y desarrollo de proyectos muy ambiciosos, que buscan modelizar el funcionamiento del sistema inmunitario humano.
5.2. Antitoxinas y toxinas: los toxoides como vacunas
Shibasaburo Kitasato (1852-1931) y Emil von Behring (1854-1917) inmunizaron cobayas con toxina diftérica inactivada térmicamente no patógena, pero muy inmunógena. Demostraron que el suero de las cobayas contenía una sustancia que evitaba los efectos nocivos de Corynebacterum diphtheriae y de su toxina, cuando las cobayas se exponían, de nuevo, a dosis letales del bacterias y toxinas. También demostraron que podían curar la difteria en un animal inyectándole el suero de un animal inmunizado. Denominaron antitoxina a la sustancia y sueroterapia a su tratamiento. Plantearon que se requería inmunizar a animales grandes, como caballos y ovejas, para lograr producir suficiente antitoxina que permitiera proteger a los humanos.
Una circunstancia que conviene resaltar es que, en vacunología, las vacunas más eficaces y que hoy se encuentran en uso, son aquéllas en las que la patogenicidad e inmunidad coinciden, tal como sucede con las antitoxinas en enfermedades toxigénicas (difteria, tétanos...).
5.3. Primeras vacunas combinadas: DTP (difteria, tétanos y tosferina). (Eng: DTaP, diphtheria, tetanus, and pertussis or whooping cough); Fr (diphtérie, tétanos et coqueluche)
Las vacunas combinadas son aquellas en las que dos o más vacunas se administran de forma conjunta. La primera, autorizada para su uso pediátrico, lo fue en 1947. Estaba compuesta por toxoides contra la difteria y tétanos. En 1949, se combinaron células enteras de tosferina a esta mezcla, creándose la DTP. En la actualidad la vacuna contra difteria, tétanos y tosferina, combina toxoides tetánico y diftérico y un componente acelular contra la tosferina (DTPa), menos reactógeno.
A partir de DTP se sigue buscando la mejor combinación que pueda ser administrada de forma conjunta. El beneficio de estas formulaciones es indudable al reducir el número de visitas e inyecciones, permitir obtener y mantener mejores coberturas vacunales y aumentar la efectividad de los programas de vacunación. Paso previo a la licencia en uso combinado se requiere asegurar que la respuesta inmunitaria a la combinación de antígenos no es inferior a la respuesta individual a cada una de las vacunas y, que el número de reacciones adversas no es superior a cuando se administran de forma individual.
Actualmente disponemos de vacunas que combinan protección frente a varias enfermedades infecciosas. Existen composiciones DTPa + Hep. B + VPI (difteria, tétanos, tosferina o pertussis, hepatitis B y poliomielitis); SRP + varicela (sarampión, rubeola, paperas y varicela); DTPa + VPI (difteria, tétanos, tosferina o pertussis y poliomielitis); DTPa + VPI + Hib (difteria, tétanos, tosferina o pertussis, poliomielitis y Hib (Haemophilus influenzae tipo b) o DTPa + Hep. B + VPI.
5.4. Vacuna de la poliomielitis: de la vacuna inactivada de Salk a la atenuada de Sabin
El virus de la poliomielitis ha sido responsable de numerosos casos de parálisis y muerte en todo el mundo. Se han identificado tres cepas de virus naturales de la poliomielitis con características inmunológicas distintas. El tipo 1 es el que más parálisis produce y suele ser el causante, más frecuente, de epidemias. Los humanos representan su único huésped natural. Es un poliovirus altamente transmisible, de persona a persona, por vía fecal-oral y respiratoria.
En sus inicios, la poliomielitis afectaba principalmente a los niños, por lo que se definió como una parálisis espinal infantil causada por un enterovirus. En 1916, un brote en Estados Unidos, le confirió un carácter epidémico con afectación de adolescentes y adultos. En 1908 los investigadores inyectaron, en Macacus rhesus, una suspensión de la médula espinal de un enfermo, demostrando su propagación por un virus. Observaron que estos macacos presentaban lesiones en la médula espinal, similares a las identificadas en los humanos, y desarrollaban parálisis en ambas piernas. Años más tarde, en 1938, se identificaron poliovirus en las heces de los pacientes y de sus contactos sanos. El análisis de las aguas residuales de Nueva York llevó a estimar una relación de 1:100 infecciones subclínicas por caso de polio. Considerando que existía una infección generalizada, la vacunación masiva de la población se consideró la estrategia adecuada para hacer frente a la propagación de la enfermedad.
La poliomielitis ha sido eliminada, en la mayoría de los países, gracias a los dos tipos de vacunas desarrolladas en la década de 1950. La primera, desarrollada por Jonas Salk y colaboradores, consistió en administrar los tres poliovirus inactivados, mediante inyección intramuscular, y se denomina VPI. En la década de 1940 se lograron cepas atenuadas tras pasar, repetidamente, el virus a través de roedores y, posteriormente, de cultivos celulares perdiendo su neurovirulencia. La cepa debilitada se logró purificar y producir, en grandes cantidades. En la década de 1950 fueron muchos los investigadores que trabajaron, en lograr una vacuna viva atenuada contra la poliomielitis. Entre otros se encontraban los grupos dirigidos por Hilary Koprowski, Herald Cox y Sabin. La vacuna desarrollada por Albert Sabin contiene los tres poliovirus atenuados, se administra por vía oral y, se denomina VPO. Cuando, en 1959, las cepas de Sabin fueron elegidas como las más seguras, ya se habían administrado millones de dosis de distintas vacunas experimentales en todo el mundo.
Muy ocasionalmente, la vacuna viva podría causar poliomielitis en personas vacunadas, al revertir las mutaciones que condujeron a su atenuación. En los países donde se usan las vacunas orales, se ha estimado un riesgo de polio por el virus derivado de la vacuna en menos de 4 casos por cada millón de nacimientos. Por ello, a medida que los países alcanzaron un buen control del poliovirus salvaje, se ha ido sustituyendo por versiones mejoradas de la vacuna VPI. Los países donde la poliomielitis sigue siendo endémica continúan utilizando la vacuna oral viva, debido a su mayor capacidad para inducir inmunidad en la mucosa intestinal, su bajo coste, la facilidad de administración y su efecto de vacunación en cadena, al eliminarse en heces y así inmunizar a personas que no hayan sido vacunadas directamente.
El saneamiento deficiente, la falta de infraestructuras de cuidados y la oposición a las campañas de vacunación, por parte de organizaciones integristas islámicas, han obstaculizado los esfuerzos de la Global Polio Eradication Initiative (GPEI) para la erradicación mundial de la poliomielitis. Los poliovirus 2 y 3 (VP2 y VP3) se erradicaron en los años 2015 y 2019. Hoy, la cepa natural de tipo 1 se encuentra circulante, exclusivamente, en dos países endémicos: Afganistán y Pakistán, la cual sufrió un aumento considerable en 2024. En el año 2022, la novedad fue el registro de una infección en un adulto en Estados Unidos y, la publicación por parte del European Centre for Disease Prevention and Control (ECDC), de la sospecha de transmisión local en Reino Unido. La respuesta rápida de las autoridades británicas del National Health Service (NHS), y sólo aplicable para Londres, consistió en poner en marcha una campaña para inocular dosis de refuerzo, a un millón de niños de entre uno y nueve años, independientemente de su estatus vacunal. El objetivo de esta acción era ampliar la cobertura vacunal, completar la protección en los no vacunados y reforzarla en los que la hubieran recibido.
En España, desde el año 2004, sólo se emplea la vacuna inactivada VPI que va incluida en los preparados de vacunas combinadas junto a otros antígenos. El porcentaje de niños primovacunados (al menos, con dos dosis) se acerca al 98 % según datos del Sistema de Información de Vacunaciones del Ministerio de Sanidad (SIVAMIN).
5.5. Otras vacunas víricas: vacuna triple vírica. Sarampión, rubeola, paperas (SRP). Eng: measles, mumps, rubella (MMR); Fr: rougeole, oreillons, rubéole (ROR)
Otras vacunas de virus vivos atenuados también han resultado importantes para la salud de la población. El virus del sarampión fue aislado, por John F. Enders, en 1954. Años más tarde, Enders y colegas elaboraron una vacuna atenuada contra el sarampión Edmonston B. Esta, junto a una segunda vacuna basada en un virus inactivado, obtuvieron, en 1963, la autorización de la U.S. Food and Drug Administration (FDA) para ser utilizadas. La FDA autorizó, en 1967, la primera vacuna contra las paperas desarrollada por Maurice Hilleman, trabajando en la compañía Merck, y basada en una muestra de virus que había aislado de su hija. En 1969 se autorizó la primera vacuna viva atenuada contra la rubeola. Posteriormente, en 1971, M. Hilleman en Merck, logró combinar las vacunas vivas atenuadas contra el sarampión, las paperas y la rubeola, MMR, en una sola inyección sin que existiera disminución de la potencia ni aumento de los efectos secundarios adversos. La rápida y generalizada aceptación de esta vacuna condujo a una caída pronunciada de la incidencia de estas enfermedades y a considerar la eliminación del sarampión en varias regiones de la OMS. Por su parte, España fue declarada por la Comisión Regional Europea de Verificación (RVC) de la OMS como país libre de transmisión endémica de sarampión el 26 de septiembre de 2017, al no haberse registrado transmisión continuada del virus entre 2014 y 2016; sin embargo, en septiembre de 2025 perdió este estatus, tras constatarse la reanudación de la transmisión endémica como consecuencia del repunte de casos observado en 2024 (incrementándose aún más en 2025), circunstancia que fue comunicada oficialmente al Ministerio de Sanidad el 27 de enero de 2026 por el RVC.
El calendario de inmunización vigente (AEP 2026) contempla dos dosis de vacuna frente a sarampión, rubeola y parotiditis (SRP). La primera dosis se administra a los 12 meses y la segunda se ha adelantado a los 24 meses de edad, recomendándose su administración en forma de vacuna tetravírica (SRPV) para corregir precozmente posibles fallos vacunales primarios y aumentar las coberturas.
El objetivo para mantener (y recuperar) el estado de eliminación del sarampión y la rubeola es alcanzar y sostener coberturas ≥95 % con ambas dosis. En España, la cobertura nacional con la primera dosis cumple este objetivo (96,6–96,7 % en 2024 según SIVAMIN), mientras que la segunda dosis continúa por debajo del 95 % (91,7–91,8 % en 2024), con variabilidad entre comunidades autónomas.
6. VACUNAS PRODUCIDAS POR ADN RECOMBINANTE: HEPATITIS B. (eng: hepatitis B; fr: hépatite B)
La hepatitis B es una enfermedad infecciosa de transmisión fundamentalmente sexual y sanguínea, causada por el virus de la hepatitis B (VHB). Este es un virus ADN que contiene numerosos componentes antigénicos como el antígeno de superficie (HBsAg), el antígeno core (HBcAg) y el antígeno e (HBeAg). La primera vacuna de la hepatitis se aprobó para uso humano en 1986, representando la culminación de las investigaciones iniciadas en 1979, sobre la clonación de antígenos del VHB, en levaduras. Fue la primera vacuna que se produjo utilizando tecnología de ADN recombinante y, aunque fue el tercer producto recombinante aprobado para uso clínico, es el más complejo en la formación de estructuras, semejantes a las partículas de virus infectivas y, en su inmunogenicidad sin causar la enfermedad.
En 1979 se secuenció el ADN viral de doble cadena y se localizó el gen S cuya secuencia codificaba para una sola proteína globular, el antígeno HBsAg. Se identificaron tres sitios potenciales de glicosilación. En 1982, se clonó el gen S en vectores de expresión de levadura. Utilizaron un plásmido que localizaba la secuencia codificante, bajo el control de un promotor de levadura constitutivo, permitiendo generar un alto nivel de HBsAg, verificado mediante inmunoensayos. Los experimentos de sedimentación y microscopía electrónica mostraron que las partículas de 22 nm eran la forma predominante de HBsAg secretada por las células de levadura transformadas, similares a las células humanas infectadas con virus. Además, al igual que las partículas de HBsAg de 22 nm de células humanas, que previamente habían demostrado ser aproximadamente 1000 veces más inmunogénicas que la proteína HBsAg sin ensamblar, las partículas generadas por levaduras fueron reconocidas por los anticuerpos específicos de HBsAg conocidos en ese momento.
La capacidad de producir HBsAg inmunogénico, en partículas similares a virus sin genoma (Virus Like Particles, VLP), representó un avance importante en vacunología. No sólo permitió la producción de vacunas contra el VHB, incapaces de infectar las células huésped, también creó un modelo para vacunas contra otros patógenos, como el virus del papiloma humano y la vacuna del paludismo, permitiendo demostrar que se podía producir una vacuna sin el propio patógeno causante de la enfermedad. Las VLP han resultado útiles en otras aplicaciones como el descubrimiento de anticuerpos, la bioimagen y la selección de células. La tecnología del ADN recombinante ha estado a la altura de su potencial para transformar la investigación básica en investigación aplicada, facilitando que una célula viva pudiera reducirse a una máquina de procesamiento de información y que la ingeniería genética se convirtiera en una parte integral de ambos ángulos de investigación.
Las vacunas de subunidades obtenidas por ADN recombinante, cuando son eficaces, presentan numerosos beneficios con respecto a las vacunas tradicionales. Dadas las ventajas de las vacunas producidas por ingeniería genética y el avance que han supuesto en el campo de la vacunología nos podríamos preguntar ¿por qué las vacunas de subunidades por ADN recombinante no han sustituido a las tradicionales? Entre los motivos podemos destacar: razones económicas, así encontramos por ejemplo, que el toxoide inactivado químicamente de difteria, es muy efectivo y no causa efectos adversos mayores- por lo que la industria no está demasiado interesada en invertir en el desarrollo de vacuna, basada en ADN recombinante, para sustituir a la actual; razones científicas y problemas técnicos por la baja expresión de determinados genes; así como dificultades con las proteínas que se pliegan de forma diferente, en células que no son de mamífero, que no se producen en la cantidad adecuada o que requieren procesamientos postraslacionales.
Actualmente la cobertura vacunal con 3 dosis de la hepatitis B alcanza al 84 % de la población mundial. La utilización generalizada del componente antihepatitis en la composición de vacunas combinadas ha llevado a una reducción drástica de la incidencia del cáncer de hígado causado por el VHB.
7. VACUNAS CONJUGADAS DE POLISACÁRIDOS
En la década de los 80, antes de la introducción de vacunas efectivas, H. influenzae tipo b (Hib) era la principal causa de enfermedad bacteriana invasora en la infancia. Neisseria meningitidis (meningococo) y Streptococcus pneumoniae (neumococo), también causan infecciones bacterianas invasoras graves, incluidas la meningitis bacteriana y la neumonía. Estas bacterias contienen azúcares en su cápsula externa, importantes para conferir inmunidad (polisacáridos). Estos polisacáridos son difíciles de ser reconocidos por el sistema inmune en los menores de 2 años, que tienen un sistema inmunitario inmaduro, y no estimulan la memoria inmunológica.
La vacuna contra Hib fue la primera de una nueva generación de vacunas de proteína-polisacárido que aumentan la inmunogenicidad de los polisacáridos bacterianos por conjugación con una proteína portadora encapsulada en polisacárido.
A finales de la década de 1960, dos grupos de investigación emprendieron la estrategia inusual de centrarse en la cápsula de polisacárido que cubre la superficie de Hib, estructura que brinda protección contra las respuestas inmunitarias del huésped y, es un factor de virulencia importante. Era conocido que el desarrollo de anticuerpos contra la cápsula resultaba crucial para adquirir inmunidad contra Hib y, postularon que esta cápsula de polisacárido podría aprovecharse como vacuna. Este enfoque difería notablemente de otras estrategias de vacunas que se centraban, principalmente, en el uso de bacterias enteras. Las vacunas de polisacáridos puros no fueron efectivas en niños menores de 18 meses, el grupo de edad con mayor riesgo de enfermedad; no logrando inducir memoria inmunológica a ninguna edad, debido a la naturaleza independiente de las células T de la respuesta al antígeno polisacárido.
Para superar este problema ambos grupos desarrollaron, de forma independiente, un método para mejorar su inmunogenicidad. Al conjugar el polisacárido, con una proteína transportadora con fuertes propiedades antigénicas, obtuvieron el primer conjugado de proteína y polisacárido. En particular, tales vacunas podrían inducir características de inmunidad humoral dependiente de células T, incluida una respuesta de memoria a las dosis de refuerzo de la vacuna. La primera vacuna conjugada aprobada, consistía en el polisacárido conjugado con toxoide diftérico (PRP-D). Esta vacuna recibió la aprobación de la FDA en 1987. Otras vacunas conjugadas más efectivas utilizan diferentes proteínas transportadoras [proteína de membrana externa meningocócica (PRP-OMP), CRM197 (PRP-CRM) y toxoide tetánico (PRP-T) han ido reemplazando a PRP-D, lo que llevó a su retirada del mercado en 2000. Las vacunas conjugadas de PRP forman parte de los calendarios sistemáticos de inmunización en numerosos países del mundo.
El éxito de estas vacunas inspiró el desarrollo de nuevas vacunas conjugadas dirigidas a bacterias encapsuladas con polisacáridos como S. pneumoniae y N. meningitidis; a un renacimiento en el descubrimiento de vacunas; y a un cambio rápido en la prevalencia y epidemiología de las enfermedades infantiles.
8. VACUNOLOGÍA REVERSA
De la bacteria Neisseria meningitidis, causante de la enfermedad meningocócica, se han descrito 12 serogrupos de los que seis -A, B, C, W, X e Y- causan la mayoría de los casos de enfermedad en todo el mundo. En EE. UU. se aíslan con mayor frecuencia tres serogrupos B, C e Y; en Europa los más frecuentes son los serotipos B, C, W e Y.
A finales de la década de 1990, las infecciones ocasionadas por las cepas de N. meningitidis del serogrupo B (MenB), se habían resistido al desarrollo de vacunas conjugadas. Durante los años 70 se habían desarrollado vacunas basadas en sus polisacáridos capsulares, encontrándose disponibles vacunas contra los serogrupos A, C, W e Y. Sin embargo, el polisacárido capsular de MenB mostró ser un inmunógeno deficiente, debido a su similitud con un polisacárido presente en tejido neuronal (ácido polisiálico). Además, por su amplia variación de secuencia, las proteínas antigénicas conocidas no resultaron adecuadas para el desarrollo de vacunas.
La secuenciación del genoma de N. meningitidis y la utilización de herramientas bioinformáticas y bioquímicas facilitó identificar una serie de antígenos novedosos y el primer caso de "vacunología reversa". El grupo de Rino Rappuoli y Maria Grazia Pizza, trabajando para el grupo Novartis, publicaron, en el año 2000 en Science, el genoma completo de una cepa MenB (MC58) y utilizaron la información genómica para identificar un número importante de nuevos antígenos unidos a su superficie. Representó el nacimiento de la “vacunología reversa”, un nuevo proceso para el desarrollo de vacunas, utilizando un enfoque ascendente desde el genoma hasta la vacuna.
Un total de 350 porciones de una secuencia de ADN de marco abierto de lectura ORFs (por Open Reading Frame en inglés) se amplificaron y clonaron en vectores de expresión en Escherichia coli. A continuación, las proteínas recombinantes obtenidas se purificaron e inyectaron en ratones, analizándose los sueros inmunitarios de estos ratones para detectar la presencia de anticuerpos específicos. También se analizó la actividad bactericida de los sueros inmunes, y demostraron que 28 proteínas producían anticuerpos bactericidas. Representó un gran avance ya que, hasta entonces sólo se habían identificado cinco antígenos capaces de inducir actividad bactericida en especies meningocócicas. Seguidamente analizaron siete ORF asociados con sueros que, habían resultado positivos en todos los ensayos anteriores, y los compararon con genes de otras cepas de Neisseria. Seleccionaron antígenos por su capacidad para inducir una amplia protección. Entre ellos se encontraban el antígeno de unión a heparina de Neisseria (NHBA), la proteína de unión al factor H (fHbp) y la adhesina A de Neisseria (NadA). Desarrollaron una vacuna con estos antígenos y la proteína de membrana altamente variable PorA. La vacuna 4CMenB, nombre comercial Bexsero, fue aprobada por la EMA para su utilización en Europa en 2013; posteriormente, en 2015, se introdujo en el programa de vacunación rutinaria del Reino Unido tras el acuerdo de GSK con el gobierno. Actualmente se utiliza para la inmunización de menores de 12 meses de edad y dispone de licencia de la FDA, para su utilización en Estados Unidos, desde el 2015. En España, la vacunación de MenB se incorporará en todas las CC. AA. en 2023 y, hasta final de 2024, a los 2 y 4 y 12 meses de edad.
En los últimos años, la secuenciación genómica se ha disparado y el enfoque de vacunología reversa se ha intentado aplicar a otros patógenos. Indicar que una limitación de esta técnica radica en que los antígenos no se pueden clasificar en función de su capacidad predictiva de inmunización; sin embargo, avances recientes en las tecnologías de células B y en biología estructural, facilitan una mejor caracterización de la inmunogenicidad de los antígenos que se puedan identificar mediante vacunología reversa. Por otra parte, la capacidad de generar anticuerpos monoclonales humanos a partir, principalmente, de células B de memoria y células plasmáticas ha permitido la detección de antígenos contra anticuerpos humanos, mejorando la evaluación y la priorización de epítopos bacterianos como futuros candidatos a vacunas. Estos avances han dado lugar a una nueva metodología para el diseño racional de vacunas la denominada “vacunología reversa 2.0”.
9. VACUNAS EN EL SIGLO XXI
9.1. Logros y retos
Las vacunas preventivas tradicionales, que confieren protección mediante la producción de anticuerpos, presentan un mecanismo que comprendemos bien y, se encuentran en amplio uso. En los últimos veinte años, el rápido desarrollo de la biotecnología ha facilitado la creación de una nueva generación de vacunas y anticuerpos. La utilización de técnicas de ADN recombinante, la nanotecnología y la incorporación de adyuvantes más potentes han permitido el diseño y producción de vacunas basadas en ácidos nucleicos y vectores virales.
Grupos de investigación, con el Instituto Jenner y la Universidad de Oxford a la cabeza, han trabajado en plataformas que utilizan plataformas de ARN mensajero, ampliamente desarrolladas en el contexto de la pandemia de COVID‑19, y han consolidado su aplicabilidad mediante la actualización periódica de formulaciones, la ampliación de indicaciones por grupos etarios y su extensión a nuevas dianas infecciosas, configurándose como una tecnología estructural de la vacunología contemporánea.
Asimismo, otra estrategia en el desarrollo de vacunas consiste en el uso de vectores virales recombinantes que codifican genes de otros patógenos. En sus inicios se empleó el virus vaccinia modificado de Ankara (MVA, Modified Vaccinia Ankara), un virus altamente atenuado, no replicativo en células humanas, con un perfil de seguridad favorable y con capacidad para incorporar grandes fragmentos de ADN recombinante en su genoma. Este vector se ha utilizado ampliamente en la investigación y desarrollo de vacunas frente al VIH, la tuberculosis y el paludismo (malaria), entre otras infecciones.
Posteriormente, se han empleado también vectores adenovirales, tanto derivados de adenovirus humanos como del chimpancé. El uso de adenovirus de origen no humano permite minimizar el problema de la inmunidad preexistente frente al vector, que podría disminuir la eficacia de la respuesta inmunitaria inducida por la vacuna.
Figura 47.6. Plataformas para el desarrollo de nuevas vacunas.

9.2. Vacunas contra la COVID-19. Vacunas ARNm, los vectores virales se hacen realidad
La aparición del SARS-CoV-2 y, la declaración de la COVID-19 como una emergencia de salud pública por la OMS, en enero de 2020, permitió que las plataformas de investigación de vacunas como las de ácidos nucleicos y vectores virales, que se venían investigando en los últimos 20 años, se convirtieran en una realidad.
En septiembre de 2008, Katalin Karikó y sus colegas de la Universidad de Pensilvania modificaron el ARN mensajero (ARNm) utilizando análogos de nucleósidos. Estas modificaciones estabilizaron la molécula y eliminaron su capacidad para inducir una respuesta inflamatoria, convirtiendo al ARNm en una herramienta prometedora para la vacunación.
En diciembre de 2020 y, en base a los datos de seguridad y eficacia de los ensayos clínicos fase 3, se autorizaron dos vacunas de ARNm para su uso de emergencia para la prevención de la pandemia de la COVID-19. Las vacunas volvieron a ser consideradas un elemento básico de la respuesta de salud pública, con más de 180 institutos de investigación y 100 empresas en todo el mundo involucradas en los esfuerzos para desarrollar y fabricar vacunas contra el SARS-CoV-2 utilizando nuevas tecnologías.
Con la autorización de estas vacunas se entró en la denominada quinta era de la vacunología. Su desarrollo ha permitido establecer modelos de producción acelerada, farmacovigilancia en tiempo real y adaptación antigénica frente a variantes emergentes.
9.3. Vacunación frente al VRS. La inmunización con anticuerpos, de la sueroterapia a los anticuerpos monoclonales
Anteriormente, en el apartado 5.2, indicábamos que los trabajos de S. Kitasato y E. Behring, sobre el papel y la transferencia pasiva de antitoxina diftérica, fueron la primera aportación al tratamiento de las enfermedades infecciosas y se encontraban en el origen de la inmunoprofilaxis pasiva. La sueroterapia fue utilizada, para el tratamiento de numerosas enfermedades bacterianas, hasta los años 40; viéndose que su mayor efecto se lograba cuando el tratamiento se iniciaba en las fases iniciales de la enfermedad.
En el capítulo 45 de este Manual se detalla el uso actual de las inmunoglobulinas como profilaxis pre y posexposición, así como el importante desarrollo de los anticuerpos monoclonales y su papel en la estrategia de la inmunización pasiva.
En la actualidad, la inmunización pasiva mediante anticuerpos monoclonales de acción prolongada constituye una estrategia preventiva integrada en programas de salud pública, especialmente frente al virus respiratorio sincitial (VRS) en población pediátrica y grupos vulnerables. Durante años, el palivizumab fue la principal herramienta en lactantes de alto riesgo; sin embargo, la incorporación de nirsevimab (y proximamente clesrovimab), dirigido frente a la proteína F en conformación prefusión y administrable en una única dosis por temporada, ha ampliado la protección a todos los lactantes durante su primera estación epidémica, marcando un cambio sustancial en la estrategia preventiva. De forma complementaria, la aprobación de vacunas de subunidades basadas en la proteína F prefusión para adultos mayores y su uso en embarazadas permite inducir protección directa en el adulto y transferir inmunidad pasiva al recién nacido, configurando así un enfoque integral de prevención del VRS.
9.4. Retos futuros en vacunas
En otras enfermedades infecciosas, especialmente aquellas en las que la protección depende fundamentalmente de la inmunidad celular, los mecanismos de correlación inmunológica siguen siendo menos conocidos, lo que dificulta el desarrollo vacunal.
Persisten importantes retos en la investigación, particularmente en las denominadas “enfermedades de la pobreza”, como el VIH/SIDA, la tuberculosis y el paludismo, cuya complejidad biológica y contexto epidemiológico plantean obstáculos científicos y logísticos significativos. No obstante, la investigación vacunal actual es amplia: en la actualidad se encuentran en desarrollo más de 130 candidatos vacunales frente a enfermedades infecciosas de elevada morbimortalidad, entre ellas el dengue, el virus respiratorio sincitial (VRS) y el citomegalovirus (CMV), lo que refleja un impulso sostenido en la innovación en vacunología.
Pasados 40 años de la secuenciación del virus VIH-1 aún no disponemos de una vacuna para la prevención del SIDA. La actual vacuna contra la tuberculosis BCG, a pesar de sus 100 años de historia y ser una de las vacunas más utilizadas del mundo, no protege contra las formas respiratorias de la enfermedad y la tuberculosis continúa transmitiéndose y siendo la segunda causa de mortalidad causada por enfermedades transmisibles, tras la COVID-19. La OMS recomienda la vacuna contra el paludismo (RTS,S/AS01), en una pauta de 4 dosis a partir de los 5 meses de edad, para los niños del África subsahariana; esta vacuna presenta una efectividad muy moderada y, a pesar de lo cual supone un importante paso en la lucha contra la enfermedad, la investigación de una mejor vacuna debe intensificarse. Conseguir vacunas efectivas frente a estas tres enfermedades es un enorme reto científico al que acompaña una escasa motivación por parte de la industria farmacéutica. No hay que olvidar que, el tiempo necesario para el desarrollo de estas nuevas vacunas es muy largo, costoso y que presenta un relativo interés económico para las multinacionales farmacéuticas. Estos tres elementos llevan a que, la investigación y desarrollo sea financiado y coordinado por organismos internacionales y su desarrollo clínico y regulatorio resulte muy complejo en situaciones de emergencia.
Asimismo, la investigación actual se orienta hacia el desarrollo de vacunas multivalentes de nueva generación, la expansión de plataformas genómicas y la integración de estrategias combinadas de inmunización activa y pasiva.
Finalmente, también conviene recordar que disponer de vacunas autorizadas y efectivas no es suficiente para lograr controlar globalmente una enfermedad. Se requiere que se cumplan otras condiciones para su mayor equidad como son el ser producidas a gran escala; un precio asequible, especialmente para los países de menores ingresos; una distribución equitativa mundial; un reparto y administración rápida, equitativa y eficiente que llegue a toda la población susceptible, especialmente, a la más vulnerable en cada país.
El control de futuras pandemias dependerá no solo de la innovación tecnológica sino también de la capacidad de producción global, la equidad en el acceso y la implementación de sistemas de decisión compartida en políticas de vacunación.
La investigación continúa, los retos son aún numerosos y mucha de la historia de las vacunas está todavía por escribir.
10. BIBLIOGRAFÍA
- Bloom DE, et al. Antimicrobial resistance and the role of vaccines. Proc Natl Acad Sci U S A. 2018;115:12868-71.
- Colditz GA, et al. Efficacy of BCG vaccine in the prevention of tuberculosis: meta-analysis of the published literature. JAMA [Internet]. 1994;271:698-702.
- Comisión Económica para América Latina y el Caribe (CEPAL). Acerca de CELADE [Internet]. Santiago de Chile: CEPAL.
- Eurostat. EU life expectancy estimated in 2024 [Internet]. Luxembourg: European Commission; 2025 Sep 11.
- Jurczak M, et al. Beyond Tuberculosis: The Surprising Immunological Benefits of the Bacillus Calmette-Guérin (BCG) Vaccine in Infectious, Auto-Immune, and Inflammatory Diseases. Pathogens. 2025;14:196.
- Ministerio de Sanidad. Portal estadístico. Área de Inteligencia de Gestión. Coberturas de vacunación en España
- Ministerio de Sanidad. Centro de Coordinación de Alertas y Emergencias Sanitarias. Informe de situación: Evaluación del Comité Regional Europeo de Verificación de la Eliminación del Sarampión y
la Rubeola. Resultados para España. [Internet]. 27 ene 2026. - Plotkin SA, et al. The development of vaccines: how the past led to the future. Nat Rev Microbiol. 2011;9:889-93.
- Querec T, et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol. 2009;10:116-25.
- Rappuoli R, et al. Medicine. The intangible value of vaccination. Science. 2002;297:937-9.
- Rappuoli R, et al. A 2020 vision for vaccines against HIV, tuberculosis and malaria. Nature. 2011;473:463-9.
- World Health Organization. TB research and innovation [Internet]. In: Global tuberculosis report 2025. Geneva: World Health Organization; 2025.
11. Enlaces de interés
- Comité Asesor de Vacunas e Inmunizaciones de la AEP. Manual de inmunizaciones en línea de la AEP. Cap. 1. Generalidades de las inmunizaciones. Tabla 1.1. Clasificación de las vacunas
- Comité Asesor de Vacunas e Inmunizaciones de la AEP. Noticias sobre historia de las vacunas
- Inmunize.org. Línea temporal de la historia de las vacunas
- Ministerio de Sanidad. Coberturas de vacunación
- Nature, 28 de septiembre de 2020. Nature Milestones in Vaccines
- The College of Physicians of Philadelphia. History of Vaccines
12. Historial de actualizaciones
| 21 de marzo de 2023 | Se crean los 12 apartados. |
| 25 de febrero de 2026 | Actualización de los 11 apartados. Nuevas citas bibliográficas y enlaces de interés |
-oOo-

