Para evitar confusiones, le informamos de que ESTE NO ES el formulario para enviar PREGUNTAS SOBRE VACUNAS al comité. Si esa fuera su intención, diríjase a la sección de preguntas de profesionales o de preguntas de familias.
A través de este formulario, el responsable del mismo, la Asociación Española de Pediatría (AEP) y su Comité Asesor de Vacunas, recaba los datos necesarios para gestionar el envío de sus comentarios y sugerencias sobre el Manual de Inmunizaciones en línea de la AEP, que ponemos a su disposición en nuestra página web.
Los campos marcados con asterisco son obligatorios y, sin ellos, no se tramitará su comentario o sugerencia.
Este tratamiento de datos no puede realizarse sin su consentimiento, por lo que deberá validar la casilla de protección de datos antes de enviar la consulta.
Sus datos no serán cedidos a otras entidades, ni transmitidos a otros países. Tiene derecho a acceder, rectificar y suprimir los datos, así como otros derechos, como se explica en la información común a los tratamientos que efectúa la AEP.
Puede consultar la información detallada sobre protección de datos, así como la información común a los tratamientos que efectúa la AEP.
50. Investigación en inmunizaciones
Capítulo 50 - Investigación en inmunizaciones
- Introducción. ¿Por qué investigamos y por qué es necesaria la investigación?
- El ensayo clínico aleatorizado (ECA) como paradigma de la evidencia científica
2.1. Características generales de un ECA
2.2. Clasificación de los ECA
3.1. Códigos de ética: las normas de buena práctica clínica (BPC) de la International Conference on Harmonisation (ICH)
3.2. Personas y entidades implicadas en el desarrollo de un ECA en vacunas
3.2.1. Autoridad regulatoria de fármacos
3.2.2. Promotor
3.2.3. Investigador
3.2.4. Comité de ética
3.2.5. Participantes en el ensayo
4.1.1. Elementos clave
4.1.2. Documentación
4.2.1. Aplicación en ECA de vacunas en menores
4.2.2. Importancia ética y legal
5.1. Fases de un ensayo clínico en investigación en vacunas
5.1.1. Estudios fase I
5.1.2. Estudios fase II
5.1.3. Estudios fase III
5.1.4. Estudios fase IV
- Bibliografía
- Enlaces de interés
- Historial de actualizaciones
- Tabla y figura incluidas en el capítulo:
Tabla 50.1. Resumen de las características de las diferentes fases del ensayo clínico en vacunas.
Figura 50.1. Diseño general del proceso de investigación.
Sugerencia para la citación: Comité Asesor de Vacunas (CAV-AEP). Investigación en vacunas. Manual de inmunizaciones en línea de la AEP [Internet]. Madrid: AEP; ago/2023. [consultado el dd/mmm/aaaa]. Disponible en: http://vacunasaep.org/documentos/manual/cap-50
1. Introducción. ¿Por qué investigamos y por qué es necesaria la investigación?
Investigar supone un proceso sistemático de descripción, recopilación y análisis de datos, con el objeto de responder a una pregunta o dar solución a un determinado problema, a través de la aplicación del método científico.
El ser humano, desde el principio de los tiempos, ha intentado comprender el comportamiento del mundo que nos rodea a través de su observación, buscando una explicación con la intención de poder predecir los acontecimientos, generándose el concepto de “explicación científica”. Sin embargo, el desarrollo de una metodología para poder obtener esta “explicación” de forma sistematizada, que en la actualidad conocemos como “método científico” y que en nuestro ámbito conocemos como “método empírico-analítico”, ha sido objeto de intenso y recurrente debate a lo largo de los últimos 3 siglos.
Desde la descripción realizada por Aristóteles de los diferentes tipos de razonamientos y la construcción del empirismo hasta la actualidad, muchos pensadores han intentado dilucidar la mejor manera de obtener el máximo conocimiento de la naturaleza que nos rodea, intentando acercar el método científico al estudio de la medicina.
Aunque resulte sorprendente, y a pesar de que todos los descubrimientos científicos son fruto de la aplicación de este método, no fue hasta mediados del siglo XX cuando la medicina basada en pruebas o en la evidencia (MBE) irrumpió en la práctica habitual de la medicina con el objetivo de mejorar la calidad de la práctica clínica y disminuir la enorme variabilidad de la práctica médica que había hasta el momento.
Para poder decidir si aplicar o no una determinada intervención es fundamental poder aplicar una jerarquización a la evidencia científica disponible. La primera disponible fue elaborada por la Canadian Task Force on the Periodic Health Examination en 1979 y adaptada después por la U. S. Preventive Services Task Force (USPSTF) en 1984. Esta jerarquía establece niveles de evidencia (NE) y grados de recomendación (GR) para sujetos asintomáticos, indicando en base a ellos qué procedimientos serían adecuados y cuáles debían ser evitados.
Desde entonces se han elaborado diferentes propuestas y clasificaciones, y todas ellas comparten como paradigma de la evidencia un tipo de estudio: el ensayo clínico.
2. El ensayo clínico aleatorizado (ECA) como paradigma de la evidencia científica
El ensayo clínico es un tipo de estudio experimental, de carácter prospectivo y controlado, de modo que el investigador “controla” las variables del estudio.
El Real Decreto de 16 de febrero de 2004 por el que se regulan en España los ensayos clínicos con medicamentos, define estos como: “toda investigación efectuada en seres humanos para determinar o confirmar los efectos clínicos, farmacológicos y/o demás efectos farmacodinámicos, y/o de detectar las reacciones adversas, y/o de estudiar la absorción, distribución, metabolismo y excreción de uno o varios medicamentos en investigación con el fin de determinar su seguridad y/o su eficacia”.
Los sujetos participantes son asignados de forma aleatoria a las distintas intervenciones que se comparan. De este modo, se establece la posibilidad de “comparar” si una nueva intervención (p.e. una nueva vacuna) es mejor frente (grupo control) a una antigua o a “no hacer nada” (placebo). Ambos grupos son seguidos de forma concurrente durante un periodo de tiempo determinado y se comparan las diferentes respuestas obtenidas en los dos grupos del estudio.
La aleatorización favorece que los grupos de intervención y de control sean similares en todo excepto en la intervención, por lo que si se detectan diferencias en la respuesta entre los dos grupos, es más probable que se deban a la intervención en estudio.
Este tipo de estudio se piensa que presenta el menor número de errores sistemáticos o sesgos; por ello se considera que constituye la mejor prueba científica para apoyar la evidencia de eficacia de una intervención concreta. Está considerado como el diseño más idóneo para evaluar la eficacia de las intervenciones sanitarias.
2.1. Características generales de un ECA
En general, un ECA debe cumplir las siguientes condiciones:
a) Pregunta o hipótesis inicial. Los resultados del ECA deben poder responder a la pregunta o hipótesis planteada. Ésta debe estar adecuadamente estructurada y deben estar definidos la población implicada, la intervención, la forma de comparación y las características de los resultados.
b) Aleatorización. La asignación de los participantes en los grupos de intervención debe realizarse mediante un procedimiento de aleatorización que asegure que todos los sujetos tengan la misma probabilidad de acceder a cualquiera de los grupos establecidos. Además, el algoritmo o secuencia de aleatorización debe permanecer oculto para asegurar que no pueda existir interferencia en la asignación, tanto por parte del investigador como de los participantes. La aleatorización permite realizar pruebas directas de causa y efecto y construir pruebas válidas de significancia estadística. Los ensayos clínicos deben utilizar métodos verificables de aleatorización, de tal manera que después del estudio el investigador pueda demostrar que la asignación se mantuvo libre de sesgo.
c) Enmascaramiento. Las intervenciones deben estar enmascaradas (procedimiento doble ciego), de forma que ni los participantes ni los investigadores conozcan qué intervención recibe cada sujeto del estudio. Este requisito reduce el sesgo de información, porque al desconocer el tipo de intervención que ha recibido el sujeto evita que se interfiera sobre la interpretación de los resultados.
d) Seguimiento. Debe ser completo e igual en los diferentes grupos de intervención, asegurando además la comparabilidad de ambas muestras en términos de tamaño muestral para evitar interferencia con los resultados del ECA.
e) Análisis de los resultados. Los sujetos se analizan en función del grupo al que fueron inicialmente asignados (análisis por intención de tratar) y según el grupo en el que finalmente participaron (análisis por protocolo). El análisis por intención de tratar da una estimación del beneficio de una intervención más cercana a la práctica diaria (efectividad) ya que incluye todos los sujetos participantes, independientemente de que hayan podido incluirse o no en el análisis por protocolo.
2.2. Clasificación de los ECA
Según diferentes criterios, la mayoría de los ensayos clínicos aleatorizados pueden clasificarse en:
a) Estructura o tipo de intervención
- Diseño paralelo: los sujetos del estudio siguen el tratamiento al que han sido asignados al azar durante el tiempo que dure el ensayo.
- Diseño de tratamiento sucesivo: cada sujeto es asignado al azar a un grupo que sigue una secuencia de tratamiento previamente determinada, de manera que cada persona recibe más de un tratamiento.
Variantes:
• Diseño de tratamiento de remplazo. Obtiene datos sobre el efecto de cambiar un tratamiento. Los sujetos se dividen en grupos iguales.
• Diseño cruzado (Crossover). Un grupo recibe un tratamiento y el otro en orden inverso. Cada sujeto sirve como su propio control.
- Diseño de ensayos alternativos
• Diseño factorial: se pueden asignar de manera aleatoria dos o más intervenciones en forma independiente, siempre y cuando no exista una interacción; de tal manera que los sujetos pueden no recibir ninguna intervención, una de ellas o eventualmente todas.
• Ensayos de equivalencia: evalúan diferencias en tratamiento cercanas a cero y con un estrecho intervalo de confianza. Se ponen en práctica porque existen tratamientos que pueden diferir en seguridad, efectos adversos, conveniencia de administración, costes, entre otras características; y el hecho de mostrar “equivalencia” tiene importancia para el uso subsiguiente de uno o ambos tratamientos.
• Aleatorización por conglomerados (Cluster)
b) Enfoque de enfermedad
- Ensayo de tratamiento
- Prevención primaria
- Prevención secundaria
La amplia mayoría de los ensayos clínicos realizados en el ámbito de las vacunas están encuadrados en el item de prevención primaria.
c) Tipo de asignación
- Fija
- Dinámica
- Adaptativa
d) Tamaño muestral
- Fijo
- Secuencial
e) Número de sedes
- Centro único: Es aquel realizado por un solo investigador o equipo de investigación en un centro hospitalario o extrahospitalario.
- Multicéntrico: Es aquel realizado en dos o más centros con un mismo protocolo y un coordinador que se encarga del procesamiento de todos los datos y del análisis de los resultados.
3. Ética de la investigación en los ECA
Cuando Edward Jenner aplicó por primera vez materia de una llaga de viruela vacuna de la mano de Sarah Nelmes a James Phipps, de ocho años de edad, para probar su hipótesis de que brindaba protección frente a la viruela, no cabe la menor duda de que su objetivo final era el encontrar la cura frente a una enfermedad que diezmó durante siglos a la población mundial. Aun así, en la actualidad, y desde un punto de vista ético, podríamos encontrar muchos errores en la metodología que empleó Jenner para sus estudios.
Por desgracia, la investigación clínica con seres humanos está llena de ejemplos de abusos cometidos por “científicos” amparados en la supuesta neutralidad de la ciencia. Este pasado reciente nos debe recordar que siempre debemos estar atentos a la exigencia de mecanismos de protección de las personas que participan en los ensayos clínicos.
La ética en la investigación es un campo de reciente desarrollo. Se trata de una ética aplicada o práctica, que trata de resolver problemas no meramente generales, sino también específicos, que surgen en la realización de la investigación. Exige que la práctica de la ciencia se realice conforme a principios éticos que aseguren el avance del conocimiento, la comprensión y mejora de la condición humana y el progreso de la sociedad. Se focaliza el interés en la perspectiva moral de la ejecución de la investigación, en la consideración de sus aspectos éticos, en su naturaleza y fines.
Existen 4 principios éticos básicos a respetar: el sujeto experimental debe dar su consentimiento libre a la investigación (autonomía de su voluntad), el investigador tiene derecho a decidir las condiciones en las que el sujeto participará, siempre y cuando, el procedimiento que se aplique alcance el mayor estándar ético y asegure el respeto a la dignidad del ser humano. Finalmente, los datos obtenidos sólo podrán utilizarse para el estudio, asegurando la protección de datos, privacidad y confidencialidad.
El primer conjunto de normas éticas para la investigación en seres humanos formuladas por la comunidad médica internacional fue establecido en 1964, tras la Segunda Guerra Mundial, por la Asociación Médica Mundial (AMM) en la Declaración de Helsinki. La AMM es una organización internacional que representa a los médicos y fue fundada en 1947. Se han realizado diversas actualizaciones, y la última versión fue aceptada en la 64.ª Asamblea General, en Fortaleza, Brasil, en octubre de 2013.
La declaración incluye un número de importantes códigos de práctica ética para la investigación en seres humanos. Define los principios éticos de forma breve y es un documento fundamental de guía para la investigación en seres humanos. A pesar de ello, no es un instrumento legal que vincule internacionalmente, aunque muchas de las leyes nacionales recogen estos principios éticos.
El principio básico de la declaración es el respeto por el individuo, su derecho a la autodeterminación y el derecho a tomar decisiones informadas incluyendo la participación en la investigación, tanto al inicio como durante el curso de la investigación. El deber del investigador es solamente hacia el paciente, y mientras exista necesidad de llevar a cabo una investigación, el bienestar del sujeto debe ser siempre prevalecer sobre los intereses de la ciencia o de la sociedad, y las consideraciones éticas deben venir siempre del análisis precedente de las leyes y regulaciones.
Toda revisión ética de proyectos de investigación y de la puesta en práctica de investigaciones en seres humanos debe ser evaluada teniendo en cuenta tanto los requisitos institucionales como las leyes pertinentes. Las leyes establecen normas que regulan la realización de investigación en seres humanos, por ejemplo, suponiendo un equilibrio aceptable entre los riesgos y los beneficios, u ocupándose de la privacidad, la confidencialidad y la propiedad intelectual. Las normas legales y los principios éticos no siempre coinciden, y pueden diferir ampliamente en las distintas jurisdicciones. Ninguna guía para la ética en la investigación en seres humanos puede proporcionar respuestas universales para todas las cuestiones éticas, ni reflejar la amplia diversidad de requisitos legales.
3.1. Códigos de ética: las normas de buena práctica clínica (BPC) de la International Conference on Harmonisation (ICH)
La buena práctica clínica (BPC) es un término acuñado en Estados Unidos (Good clinical practice, GCP) que engloba una serie de normas dirigidas a garantizar los derechos de los sujetos que participan en un ensayo clínico, asegurar la calidad de los datos y evitar errores en la investigación clínica.
En 1995, en la Conferencia Internacional de Armonización (International Conference on Harmonisation, ICH), la Unión Europea junto con Japón y Estados Unidos consiguió llegar a consensuar una guía común de normas de BPC que deben cumplir los ensayos clínicos que se presenten como base para la autorización de medicamentos en dichas áreas geográficas. Este documento de consenso fue aprobado en 1996 por el Comité de Medicamentos de Uso Humano (actual Committee for Medicinal Products for Human Use, CHMP) de la Agencia Europea de Medicamentos (European Medicines Agency, EMA) y entró en vigor en 1997.
De acuerdo con dicho documento, titulado "Normas de BPC" (Note for guidance on Good Clinical Practice, CPMP/ICH/135/95), la BPC se define como una norma internacional de calidad científica y ética dirigida al diseño, registro y redacción de informes de los ensayos clínicos en los que participan seres humanos.
El cumplimiento de estas normas garantiza públicamente la protección de los derechos, seguridad y bienestar de los sujetos que participan en el estudio y asegura la integridad y credibilidad de los datos obtenidos en un ensayo clínico. El objetivo de la armonización es eliminar las demoras innecesarias en el desarrollo y la disponibilidad mundial de nuevos medicamentos, y mantener al mismo tiempo las garantías de calidad, inocuidad y eficacia, y las obligaciones regulatorias para proteger la salud pública. Hasta el momento sólo existe una versión de la IHC BPC, la versión original publicada en 1997.
“Las buenas prácticas clínicas (BPC) son un estándar de calidad ética y científica internacional para el diseño, la realización, el registro y la comunicación de ensayos en los que participan seres humanos. El cumplimiento de ese estándar proporciona una garantía pública de que los derechos, la seguridad y el bienestar de los participantes de los ensayos están protegidos, de manera coherente con los principios originados en la Declaración de Helsinki, y que los datos del ensayo clínico son creíbles”
La BPC se ha convertido en la norma principal a nivel internacional para la realización de ensayos clínicos. No es tanto un documento sobre políticas, sino más bien una guía operativa, que describe las responsabilidades y los asuntos operativos relacionados con los ensayos clínicos.
La Directiva Europea 2001/20/EC puso las bases legales y administrativas para la aplicación de las normas de BPC en los ensayos clínicos con medicamentos realizados en Europa. Esta Directiva ha sido transpuesta en España mediante el Real Decreto 223/2004, de 6 de febrero, que regula los ensayos clínicos con medicamentos.
La Directiva 2005/28/EC establece los principios y directrices detalladas de la BPC respecto a los medicamentos en investigación de uso humano, así como los requisitos para autorizar la fabricación o importación de dichos medicamentos. Esta directiva ha sido transpuesta en España por medio de la Orden SCO/256/2007 de 5 de febrero, modificada por la Orden SCO/362/2008.
Los ensayos clínicos en los que se basa la autorización de comercialización de los medicamentos en Europa deben seguir las normas de BPC de acuerdo con la Directiva 2004/27/EC transpuesta en España mediante el Real Decreto 1345/2007, de 11 de octubre, que regula la autorización, registro y dispensación de los medicamentos de uso humano.
Todas las personas y entidades implicados en el desarrollo de un ensayo clínico deben tener en cuenta, no solo las normas de BPC, sino también las directrices científicas sobre calidad, seguridad y eficacia de los medicamentos de uso humano adoptadas por el CHMP y publicadas por la EMA, así como las demás directrices farmacéuticas comunitarias publicadas por la Comisión Europea en los distintos volúmenes de las Normas sobre Medicamentos en la Unión Europea.
Ministerio de Sanidad (Normas de Buena Práctica Clínica)
3.2. Personas y entidades implicados en el desarrollo de un ECA en vacunas
Fundamentalmente, el ámbito de un ensayo clínico esta formado por las siguientes entidades y personas: la autoridad regulatoria de fármacos, el promotor del ensayo, las empresas de gestión y monitorización (Contract Research Organization, CRO) el investigador clínico y el comité de ética (CE).
El desarrollo de una nueva vacuna está formado por un patrón estricto de interacción. Por ello, están definidas las diferentes responsabilidades que permitan el desarrollo de ensayos clínicos de alta calidad, de manera segura y ética.
El promotor interactúa tanto con la autoridad regulatoria como con el investigador antes, durante y después del ensayo, mientras que el investigador interactúa con el CE generalmente.
No obstante, el personaje fundamental de un ECA son los participantes del ensayo, pacientes o voluntarios sanos. Aunque no participen en el ensayo clínico a través del desarrollo o la planificación del ensayo, sin ellos sería completamente imposible llevar a cabo la investigación.
3.2.1. Autoridad regulatoria de fármacos
Las autoridades regulatorias de fármacos son las responsables de la garantía de la calidad en el desarrollo de nuevas vacunas, al igual que en la producción, la distribución, el etiquetado y la monitorización de la seguridad.
Cada país tiene su propia autoridad regulatoria de fármacos. Estas autoridades elaboran normas para la aprobación y realización de protocolos de ensayos clínicos. Si el ECA se realiza en diferentes países, puede ser supervisado por varias autoridades regulatorias de fármacos en función del país.
Las autoridades regulatorias de fármacos reciben distintos nombres en los diferentes países. Por ejemplo, en los EE. UU. la autoridad es la Administración de Alimentos y Medicamentos (FDA), en la Unión Europea se llama Agencia Europea de Medicamentos (EMEA); y en Japón, es el Ministerio de Salud, Trabajo y Bienestar Social (MHLW). Otros ejemplos son, Salud Canadá (Canadá), la Administración de Alimentos y Medicamentos del Estado, (SFDA, China), la Administración de Bienes Terapéuticos (TGA, Australia), el General de Control de Fármacos de la India (DGCI, India), la Agencia Nacional de Vigilancia Sanitaria (ANVISA, Brasil), y el Servicio Federal de Control de la Atención Médica y el Desarrollo Social (Roszdravnadzor; Rusia).
Son responsabilidades de las autoridades regulatorias, entre otras, revisar y aprobar los protocolos de ensayos clínicos y asegurar que los ensayos clínicos cumplan con las normas nacionales de un país y las directrices internacionales.
3.2.2. Promotor
El promotor de un ensayo clínico es una persona, empresa, institución u organización que se responsabiliza de la gestión y la financiación de un ensayo clínico.
Un promotor puede ser una empresa farmacéutica o biotecnológica, una organización sin fines de lucro como un fondo de investigación, una organización gubernamental o una institución en la que se llevará a cabo el ensayo o un investigador individual.
El promotor solicita el permiso de la/s autoridad/es reguladora/s para el inicio del ensayo, presentando un protocolo del ensayo clínico que debe ser evaluado y aprobado por un comité ético de ensayos clínicos. Debe asegurar, además, la coordinación entre las actividades de los ensayos clínicos mientras dure el estudio (desarrollo del protocolo, solicitudes regulatorias, auditorias, gestión de los datos clínicos, pruebas de laboratorio, etc). Debe asegurar también que los investigadores dispongan de información completa y actualizada acerca del estudio.
La monitorización de ensayos clínicos es uno de los procesos clave para lograr el éxito en una investigación. El monitor trabaja para asegurar que en los ensayos se obtienen datos fiables, respetando la normativa y la ética. El promotor debe asegurar la existencia de monitores o asociados de investigación clínica que actúen en su representación. El monitor es el enlace entre el promotor y el investigador clínico y su equipo de investigación, interactuando regularmente, monitoreando los diferentes procesos y el cumplimiento del protocolo.
3.2.3. Investigador
Los equipos de investigación están formados habitualmente por el investigador principal y uno o varios subinvestigadores, uno o varios enfermeros y, en caso de ser necesario, demás personal de apoyo para el estudio. Puede pertenecer a centros médicos académicos, hospitales públicos o centros de atención primaria, organizaciones médica privadas o instalaciones de investigación.
El investigador principal es el responsable de la conducción del ensayo clínico en el lugar en el que se lleva a cabo y del equipo de personas a su cargo. El subinvestigador es cualquier miembro del equipo del ensayo clínico designado y supervisado por el investigador principal. El coordinador del estudio se ocupa de las responsabilidades administrativas revisando toda la información y los registros antes de la visita de un monitor. Es responsabilidad del investigador, entre otras, proteger los derechos y el bienestar de los participantes, cumplir con las normas de Buena Práctica Clínica (BPC) y protocolo, asegurarse de que el ensayo clínico se evalúe por un comité ético e informarle en el caso en el que se produzca una desviación del protocolo o cualquier evento adverso, asegurar el proceso de consentimiento/asentimiento informado de los participantes y respetar y proteger la identidad de los participantes.
3.2.4. Comité de ética
El CE de ensayos clínicos actúa, siguiendo las normas de buena práctica clínica (BPC), asegurando la protección de los derechos, la seguridad y el bienestar de los participantes en el ensayo clínico. Presta especial atención a los ensayos que puedan incluir participantes vulnerables, como pacientes pediátricos. El CE proporciona una garantía pública de esa protección, evaluando y aprobando o rechazando el protocolo y asegurando que el investigador o los investigadores sean aptos para la conducción del ensayo, que se disponga de las instalaciones adecuadas y que los métodos y materiales que se utilizarán para obtener y documentar el consentimiento informado de los participantes en el ensayo sean apropiados. Así mismo, se encarga de revisar eventos adversos y cualquier perjuicio que ocurra como resultado del ensayo.
La condición legal, la composición, la función, el funcionamiento y los requisitos regulatorios pertenecientes a los CE independientes difieren según el país. Los miembros del CE deben representar tanto a la institución que desarrolla el ensayo como a los participantes en el mismo. Además, en el ámbito científico, deben representar diversidad de conocimientos (médicos, epidemiológicos, estadísticos,…) y experiencia para asegurar una revisión exhaustiva del ECA.
3.2.5. Participantes en el ensayo
Los ensayos clínicos en vacunas se realizan en participantes o voluntarios generalmente sanos. Los participantes generalmente se seleccionan del grupo común de pacientes del lugar del ensayo, pero también pueden proceder derivados de otras clínicas o a través de anuncios locales. La participación en los ensayos es voluntaria y altruista.
4. Consentimiento y asentimiento informado en ECA de vacunas
Los ensayos clínicos desempeñan un papel crucial en el desarrollo y la evaluación de nuevas vacunas, siendo esenciales para garantizar su seguridad y eficacia antes de su aprobación y distribución masiva.
El consentimiento y el asentimiento informado son componentes éticos fundamentales en la realización de estos ensayos clínicos.
4.1. Consentimiento Informado
El consentimiento informado es un proceso por el cual un individuo voluntario y competente, que participará en un ensayo clínico de vacunas, recibe información completa y comprensible sobre los riesgos, beneficios y procedimientos del estudio antes de tomar una decisión informada de participar o no.
Es un principio ético y legal fundamental que garantiza que los participantes en los ensayos clínicos comprendan plenamente la naturaleza del estudio y puedan tomar una decisión informada y autónoma sobre su participación.
En el contexto de los ensayos clínicos de vacunas, el consentimiento informado adquiere una importancia aún mayor debido a la naturaleza de estos estudios, que se realizan sobre personas sanas. Los ensayos clínicos de vacunas son cruciales para evaluar la seguridad y eficacia de nuevas vacunas antes de que estén disponibles para su uso generalizado. Estos ensayos implican la participación de voluntarios sanos, quienes, al recibir la vacuna en desarrollo, asumen ciertos riesgos en beneficio de la sociedad en general.
El consentimiento informado en los ensayos clínicos de vacunas se basa en la premisa de que los participantes deben recibir información completa y comprensible sobre el estudio antes de decidir participar. Esto incluye detalles sobre el propósito del ensayo, el diseño de la investigación, los procedimientos involucrados, los posibles efectos secundarios, los beneficios esperados y cualquier alternativa de tratamiento disponible. Además, los participantes deben recibir información sobre sus derechos, incluida la capacidad de retirar su consentimiento en cualquier momento sin repercusiones negativas.
El proceso de consentimiento informado se lleva a cabo mediante una comunicación clara y efectiva entre los investigadores y los participantes potenciales. Los investigadores deben proporcionar información escrita y verbal de manera comprensible, respondiendo a todas las preguntas y aclarando cualquier duda que pueda surgir. También es esencial garantizar que los participantes tengan suficiente tiempo para reflexionar sobre la información proporcionada antes de tomar una decisión.
Al implementar el consentimiento informado en los ensayos clínicos de vacunas, se deben seguir ciertos principios éticos y legales.
En primer lugar, el consentimiento debe ser voluntario, lo que significa que los participantes no pueden ser presionados ni coaccionados para participar.
Además, se debe garantizar la confidencialidad de los datos personales de los participantes y se deben obtener los permisos necesarios para recopilar, almacenar y utilizar la información relacionada con el estudio.
4.1.1. Elementos Clave
Existen varios elementos clave que deben abordarse en el proceso de consentimiento informado en los ensayos clínicos de vacunas. Estos incluyen:
- Información completa: Los participantes deben recibir información detallada y comprensible sobre el propósito del estudio, los procedimientos que se llevarán a cabo, los posibles riesgos y beneficios, así como las alternativas disponibles. Esta información debe presentarse de manera clara y en un lenguaje que sea comprensible para los participantes
- Comprensión: Los participantes deben comprender la información proporcionada y tener la oportunidad de hacer preguntas. Es fundamental evaluar la comprensión de los participantes sobre la información proporcionada. Los investigadores deben asegurarse de que los participantes entiendan los conceptos clave y puedan tomar decisiones informadas basadas en esa comprensión
- Voluntariedad: La participación debe ser completamente voluntaria, sin presiones ni coerción. Los participantes tienen el derecho de retirarse en cualquier momento sin consecuencias negativas.
- Capacidad para consentir: se debe evaluar la capacidad del participante para otorgar un consentimiento informado. Esto implica determinar si el participante tiene la capacidad de comprender la información y las implicaciones de su participación en el estudio.
- Oportunidad de hacer preguntas: los participantes deben tener la oportunidad de hacer preguntas y recibir respuestas satisfactorias antes de tomar una decisión. Esto promueve una comunicación abierta y permite que los participantes comprendan plenamente lo que implica su participación en el estudio.
El consentimiento informado es esencial en los ensayos clínicos de vacunas, pero en algunas ocasiones su aplicación puede plantear desafíos en su aplicación. Algunos participantes pueden tener dificultades para comprender la información debido a barreras idiomáticas, bajo nivel educativo o limitaciones cognitivas. En estos casos, se deben tomar medidas adicionales para garantizar la comprensión y la toma de decisiones informadas. Ningún participante que no cumpla los requisitos necesarios debe ser incluido en un ensayo clínico.
Además, la presión social o la influencia de terceros pueden afectar la capacidad de los participantes para tomar decisiones autónomas. Por lo tanto, es crucial que los investigadores brinden un entorno libre de coerción y se aseguren de que los participantes comprendan que su participación es completamente voluntaria.
4.1.2. Documentación
El consentimiento informado se documenta mediante un formulario escrito que el participante firma, pero es fundamental recordar que la firma no es el único elemento importante. La comprensión de la información aportada en el documento escrito es esencial, y los investigadores deben asegurarse de que los participantes estén realmente informados y de acuerdo con su participación.
4.2. Asentimiento Informado
El asentimiento informado es un proceso similar al consentimiento informado, pero aplicado a individuos que, debido a su edad o capacidad cognitiva, no pueden otorgar un consentimiento totalmente informado por sí mismos. Este proceso involucra la obtención del acuerdo del individuo en la medida de su capacidad, así como el consentimiento de los padres o tutores legales.
4.2.1. Aplicación en ECA de vacunas en menores
En los ensayos clínicos de vacunas que involucran a niños y adolescentes, el asentimiento informado cobra elevada importancia. Los investigadores deben explicar el estudio de manera adecuada y comprensible para la edad del niño, fomentando su participación activa en la toma de decisiones en la medida en que sea posible. Habitualmente, en España, se solicita asentimiento a todo participante de 12 a 17 años de edad.
El asentimiento informado se refiere al proceso mediante el cual un menor de edad expresa su acuerdo para participar en un ensayo clínico, después de recibir información adecuada y comprensible sobre el estudio y sus implicaciones. Aunque los menores no tienen la capacidad legal para otorgar un consentimiento pleno, se considera esencial obtener su asentimiento para respetar su autonomía y proteger sus derechos.
En los ensayos clínicos de vacunas, el asentimiento informado adquiere una relevancia particular, ya que, en muchos casos, implica la participación de personas menores de edad. Los ensayos clínicos de vacunas pediátricas son cruciales para evaluar la seguridad y eficacia de las vacunas en esta población, y es fundamental garantizar que los niños y adolescentes comprendan la naturaleza del estudio y sus posibles riesgos y beneficios.
El proceso de asentimiento informado en los ensayos clínicos de vacunas sigue principios similares al consentimiento informado de los adultos, adaptados a la edad y capacidad de comprensión de los menores.
Es esencial que los investigadores proporcionen información adecuada y comprensible a los niños y adolescentes, explicando el propósito del estudio, los procedimientos involucrados, los posibles efectos secundarios y los beneficios esperados.
Además, los investigadores deben asegurarse de que los menores comprendan la información presentada y tengan la oportunidad de hacer preguntas para aclarar sus dudas. El lenguaje utilizado debe ser adaptado a su nivel de desarrollo y se deben utilizar métodos visuales o interactivos para facilitar la comprensión.
4.2.2. Importancia ética y legal
El proceso de asentimiento informado también debe considerar el contexto familiar y social del menor. Los investigadores deben obtener el consentimiento de los padres o tutores legales, quienes son responsables de proteger los intereses y el bienestar de sus hijos. Sin embargo, el asentimiento del menor sigue siendo esencial para respetar su autonomía y participación voluntaria.
Es importante destacar que el asentimiento informado no significa que el menor tome todas las decisiones por sí mismo, sino que se le brinda la oportunidad de expresar su acuerdo y opinión. En última instancia, los padres o tutores legales tienen la responsabilidad de evaluar la información proporcionada y tomar la decisión final sobre la participación del menor en el ensayo clínico.
La implementación efectiva del asentimiento informado en los ensayos clínicos de vacunas pediátricas implica un enfoque multidisciplinario y colaborativo. Los investigadores, los profesionales de la salud y los padres o tutores deben trabajar juntos para garantizar que se respeten los derechos y la protección de los menores, al tiempo que se promueve una participación informada y ética.
5. ¿Cómo se investiga en vacunas?: el ECA aplicado al campo de la prevención mediante vacunación
Tras más de dos siglos del inicio de la primera inmunización con la vacuna de la viruela, los procedimientos de evaluación y control de estos fármacos han ido progresivamente perfeccionándose. Existe en la actualidad una legislación específica y compleja que regula, entre otras cuestiones, la investigación en este campo.
Cada nueva vacuna candidata a ser aplicada a la población debe evaluarse para determinar su seguridad, inmunogenicidad y eficacia protectora en seres humanos antes de que tenga licencia para su uso. Para ello, se utiliza como metodología de estudio el ensayo clínico, que como veíamos en apartados anteriores, se considera el paradigma actual de la evidencia.
Después de la evaluación inicial de seguridad, cada vacuna candidata sigue un camino de desarrollo único y diferente, en función de que se trate de una nueva vacuna para la prevención de una enfermedad o que se trate de un nuevo producto con diferente mecanismo de protección a los anteriores o una modificación del mismo. Influyen además en su desarrollo las características específicas de cada vacuna (organismos vivos, proteínas, subunidades…), la epidemiología de la enfermedad, el tipo de población a la que se va aplicar, etc.
El desarrollo clínico de una vacuna está basado fundamentalmente en las directrices emitidas por la Organización Mundial de la Salud (OMS), la Agencia Europea de Medicamentos (EMA) y la Administración de Fármacos y Alimentos de los Estados Unidos (US-FDA). Estas directrices incluyen los objetivos, población a estudio, diseños de estudio, lugares donde se realiza y resultados de cada una de las fases (Fase I-III) de un ensayo clínico.
Las agencias reguladoras de todo el mundo dividen este proceso de desarrollo en fases preclínicas (pruebas in vitro e in vivo en animales) y clínicas (ensayos clínicos en seres humanos).
Pocas vacunas candidatas consiguen pasar del laboratorio (fase preclínica) a la fase de ensayos clínicos. Para poder hacerlo y continuar su desarrollo, deben superar criterios como la inmunogenicidad demostrada en animales de experimentación, ausencia de toxicidad, la existencia de respuesta de inmunogenicidad, impacto en la salud pública, la rentabilidad y la preexistencia de una vacuna con un perfil de riesgo-beneficio satisfactorio.
Los ECA se consideran en la actualidad el "estándar de oro" de la investigación en vacunas. Son estudios aleatorizados en donde los participantes se asignan al azar para recibir la vacuna de investigación o el control (placebo, vacuna diferente). Su carácter prospectivo permite el control de las variables, evita el sesgo y maximiza las posibilidades de detectar una diferencia entre la vacuna en investigación y el control.
A estas cuestiones se suma una exigencia mayor y especial cuando se realizan ensayos clínicos de vacunas en una población pediátrica, a diferencia de los medicamentos que se administran a los pacientes, las vacunas son administradas a sujetos sanos, por lo que el margen de seguridad debe ser muy alto.
Como los niños sanos también reciben inmunización bajo el programa de vacunación sistemática de cada país, el diseño del ensayo se complica debido a la posibilidad de interferencia durante la coinmunización o coadministración.
Además, el desarrollo clínico en pediatría implica que previamente el producto debe haber sido evaluado en términos de seguridad primero en adultos, seguido de los adolescentes y niños antes de poder ser evaluado en lactantes.
Antes de la aprobación reglamentaria, una vacuna candidata generalmente se somete a tres fases de desarrollo en seres humanos, que, en su mayor parte, progresan secuencialmente: fase I, fase II y fase III.
Después de completar con éxito los ensayos de la fase III y luego de obtener la licencia del producto, los estudios de la fase IV, también denominados estudios de vigilancia posterior a la comercialización, se utilizan para continuar monitorizando la seguridad y la efectividad de la vacuna una vez aplicada a la población (tabla 50.1).
Tabla 50.1. Resumen de las características de las diferentes fases del ensayo clínico en vacunas.
| Tamaño muestral | Objetivo | Diseño | |
| Fase I |
Pequeño número de voluntarios sanos (20 -80 aproximadamente) |
Seguridad
Respuesta inmunológica |
No controlado |
| Fase II | Voluntarios sanos
(100-400 aproximadamente) |
Seguridad Capacidad imunógena Dosis propuestas Programa de vacunación Método de aplicación
|
Controlados Asignación aleatoria
|
| Fase III | Voluntarios sanos
(muestra de gran tamaño: “miles”, heterogénea y representativa) |
Seguridad Confirmación de la eficacia Beneficios en salud |
Controlados
Asignación aleatoria |
| Fase IV | Población general |
Efectividad Farmacovigilancia |
Larga duración |
5.1. Fases de un ensayo clínico en investigación en vacunas
5.1.1. Estudios fase I
La fase I del desarrollo de una nueva vacuna candidata tiene como objetivo principal evaluar la seguridad y la reactogenicidad del fármaco, siendo un objetivo secundario la evaluación de la respuesta inmune. En esta fase también se suele evaluar de manera inicial la dosis, la pauta de vacunación y el modo de administración de la vacuna.
Estos estudios generalmente se realizan en poblaciones de pequeño tamaño, inicialmente en adultos sanos e inmunocompetentes, aunque en alguna ocasión, por las características de la enfermedad a prevenir, puede realizarse en población infantil. Según el resultado de estos estudios (denominados ensayos fase Ia), los posteriores se pueden realizar en diferentes grupos de edad o en población más parecida a la población diana (fase Ib), que permite evaluar posibles diferencias en cuanto a dosis, seguridad, pauta o vía de administración.
En esta fase de desarrollo algunos ensayos pueden ser abiertos y no aleatorizados, en función del objetivo principal de seguridad que se busque. No obstante, la mayoría son ECA, frente a un placebo o un preparado vacunal frente a una enfermedad diferente, como comparador. Para controlar el sesgo, el estudio puede ser simple o doble ciego. Estos estudios se realizan en el ámbito hospitalario o en centros que disponen de un hospital de atención terciaria cercano.
Después de la inmunización de los pacientes es necesaria la observación con el fin de monitorizar los eventos adversos que pudieran aparecer. La tolerancia y la reactogenicidad debidas a la vacuna o al proceso de vacunación es el principal resultado de seguridad evaluado en un ensayo de fase I.
Las pruebas de laboratorio de seguridad clínica (por ejemplo, hematología, bioquímica, análisis de orina) también forman parte de los datos de seguridad que se recopilan al inicio del estudio, a intervalos definidos y al final del ensayo. Los ensayos de inmunogenicidad deben validarse y realizarse preferiblemente de acuerdo con las buenas prácticas de laboratorio clínico (GCLP).
La EMA establece una serie de directrices para la presentación de los datos inmunológicos de las vacunas, que se resumen del siguiente modo:
- Porcentaje de "respondedores" o "seroconversores" (con intervalo de confianza (IC) del 95 %). Los respondedores son individuos que desarrollan una respuesta inmune superior a un cierto nivel o umbral o aquellos que alcanzan un cierto incremento mínimo en la concentración o título de anticuerpos después de la vacunación. Estos incrementos no implican necesariamente protección y deben estar recogidos en el protocolo antes de iniciar el estudio.
- Concentración media geométrica/títulos medios geométricos (con IC del 95 %) y las proporciones previas/posteriores a la vacunación (proporciones medias geométricas) proporcionan valores absolutos y aumento en los títulos de anticuerpos en los puntos de tiempo definidos después de cada vacunación.
- Curvas de distribución acumulativa inversa (RCD) muestran el porcentaje de vacunados frente a los niveles de anticuerpos, lo que permite una comparación directa de las respuestas logradas en diferentes grupos de estudio.
- Respuestas células T específicas de antígeno, incluyendo el grupo de diferenciación (CD) 4+ y los linfocitos T citotóxicos (CTL) CD8 + y las citoquinas relevantes, si corresponde.
5.1.2. Estudios fase II
Las vacunas candidatas, tras lograr un resultado satisfactorio en los estudios de fase I en términos de seguridad e inmunogenicidad, continúan con su evaluación clínica a través de la fase II.
En esta fase el número de participantes es mucho mayor que en la fase I, pasándose de un entorno clínico controlado a una primera evaluación de campo. Se reclutan, por lo general, cientos de sujetos de la población objetivo de forma multicéntrica, lo que permitirá concluir con certidumbre si la vacuna candidata es segura, suficientemente inmunogénica y quizás protectora. Estos ensayos se realizan habitualmente en el ámbito de la atención primaria, que dispone de acceso a prácticamente toda la población general.
El objetivo de esta fase consiste sobre todo en la estimación de la dosis óptima de vacuna y la pauta vacunal óptima.
Del mismo modo que en la fase I, en la fase II se sigue evaluando la seguridad de la vacuna estudiada. Por regla general, el control utilizado en estos estudios es un placebo que permite estimar mejor la tasa de reacciones adversas detectadas. El tamaño muestral de estos estudios se calcula en función de la potencia estadística deseada y se espera que proporcionen un resultado clínicamente significativo en términos de seguridad, inmunogenicidad y eficacia.
Los estudios de fase II evalúan el impacto de múltiples variables en la respuesta inmune, como la edad, raza, género, presencia de anticuerpos preexistentes, etc, y tienen en consideración tanto la edad de administración de la vacuna, como el número y el intervalo entre dosis. Para vacunas cuya población diana son los lactantes, habitualmente se establece un proceso progresivo de estudio que se inicia en adultos, adolescentes y niños antes de realizar el ensayo en lactantes.
Así mismo, los estudios fase II se subdividen en diferentes estudios para poder estimar de forma correcta el impacto de variables como la estimación de la dosis seleccionada, el programa elegido, el grupo de edad y la duración del seguimiento, antes de continuar con los estudios de fase III. Su máximo interés es proporcionar información sobre la eficacia protectora en humanos de las dosis empleadas. Estos estudios, denominados fase IIa, hacen referencia a los estudios iniciales. Se realizan con un número delimitado de sujetos y permiten una evaluación rápida de si el preparado vacunal tiene características óptimas para seguir avanzando en la investigación y progresar hacia otros estudios, denominados fase IIb o estudios finales, previos a la fase III. Los estudios fase IIa se realizan en pocos sujetos, con criterios de inclusión/exclusión más estrictos, y los fase IIb, que incluyen más sujetos, evalúan la eficacia y la seguridad y representan una demostración más rigurosa de la eficacia del nuevo compuesto.
Del mismo modo que sucede en los estudios de fase I, en los de fase II se evalúa la respuesta humoral y la concentración mínima inhibitoria (CMI) considerada como inmunógena en la vacunas, pero en esta ocasión con el objeto de determinar la dosis óptima a emplear. En la mayoría de ocasiones, la vacuna deberá demostrar de forma directa su eficacia frente a la enfermedad en los estudios.
Este precepto se cumple para todas las vacunas con la excepción de las que protegen frente a enfermedades de muy baja incidencia aunque de elevada morbimortalidad. En este caso se admite la utilización de los correlatos serológicos de protección como medida indirecta de la eficacia vacunal. Esto es necesario porque una incidencia baja significa que se requiere un gran número de sujetos para estimar la eficacia de la vacuna en comparación con los números necesarios si la incidencia de la enfermedad es mayor. Estos correlatos de protección estimados también se emplean en la fase III. Un ejemplo de su uso son las vacunas conjugadas frente a los meningococos C o ACWY. Estas vacunas se autorizaron en España en base a estos correlatos serológicos, sin datos de eficacia directa.
Los eventos adversos detectados en la fase I (solicitados y no solicitados) son recopilados también en la fase II con el objeto de estimar la seguridad vacunal. Dado que el tamaño muestral en esta fase es mucho mayor, los estudios de la fase II tienen mayor potencia estadística para estimar diferencias significativas de los eventos adversos entre los grupos. Del mismo modo orientan con su resultado los eventos adversos específicos que deben evaluarse en ensayos de fase III con mayor tamaño muestral.
5.1.3. Estudios fase III
Los ensayos fase III son esenciales para el registro, aprobación y comercialización de una vacuna. El objetivo principal de un ensayo de fase III es demostrar o confirmar el beneficio terapéutico, la evidencia previa recopilada durante las fases previas, es decir, que la vacuna es segura y eficaz para su uso en la indicación y población específica. Esos estudios proporcionan la base para la aprobación y posterior lanzamiento al mercado.
Estos ensayos están diseñados típicamente para evaluar la eficacia y la seguridad, y se realizan a gran escala, es decir, con la participación de miles de sujetos, lo que aumenta todavía más la potencia estadística de los estudios. Se denominan también “estudios de campo”, ya que el elevado número de participantes hace que la condición se asemeje a cuando la vacuna sea utilizada de forma rutinaria en un futuro.
Los ensayos clínicos fase III también se subdividen en: estudios fase IIIa, que se llevan a cabo una vez determinada la eficacia y constituyen la evidencia de efectividad que se exige para la tramitación del expediente de la nueva vacuna. Estos estudios se denominan también “ensayos pivotales” y son fundamentales en la solicitud de valoración de la nueva vacuna por las autoridades reguladoras. La fase IIIb incluye estudios que se realizan una vez aceptada la tramitación y antes de la aprobación y comercialización de la nueva vacuna.
En los estudios fase III de vacunas frente enfermedades infecciosas se pueden evaluar diferentes objetivos en función de las prioridades para la salud pública y para la enfermedad que se desea proteger. En función de estas características, se pueden evaluar: la eficacia de la vacuna en función de la susceptibilidad, colonización, progresión y patogenicidad e infecciosidad de la enfermedad, efectos indirectos de la vacunación en los no vacunados y efectos generales a nivel de la población.
Si los resultados de la fase III demuestran eficacia y seguridad, el fabricante de la vacuna puede enviar una solicitud a la autoridad reguladora nacional para otorgar licencias y comercializar el producto.
5.1.4. Estudios fase IV
La fase IV constituye la ampliación de conocimiento sobre la eficacia de la vacuna una vez obtenida la aprobación para la comercialización y comienza a aplicarse de forma sistemática en la población. Además de las reacciones adversas que pudieran ocurrir con su uso y que no hubieran sido detectadas en las fases anteriores, se evalúa también la efectividad a través de la vigilancia epidemiológica continuada.
Las fases anteriormente descritas suelen transcurrir de forma consecutiva, aunque en ocasiones pueden superponerse entre sí, comenzando una fase posterior sin haber terminado las previas.
Un resumen de todo el proceso investigador se puede observar en la figura 50.1
Figura 50.1. Diseño general del proceso de investigación.
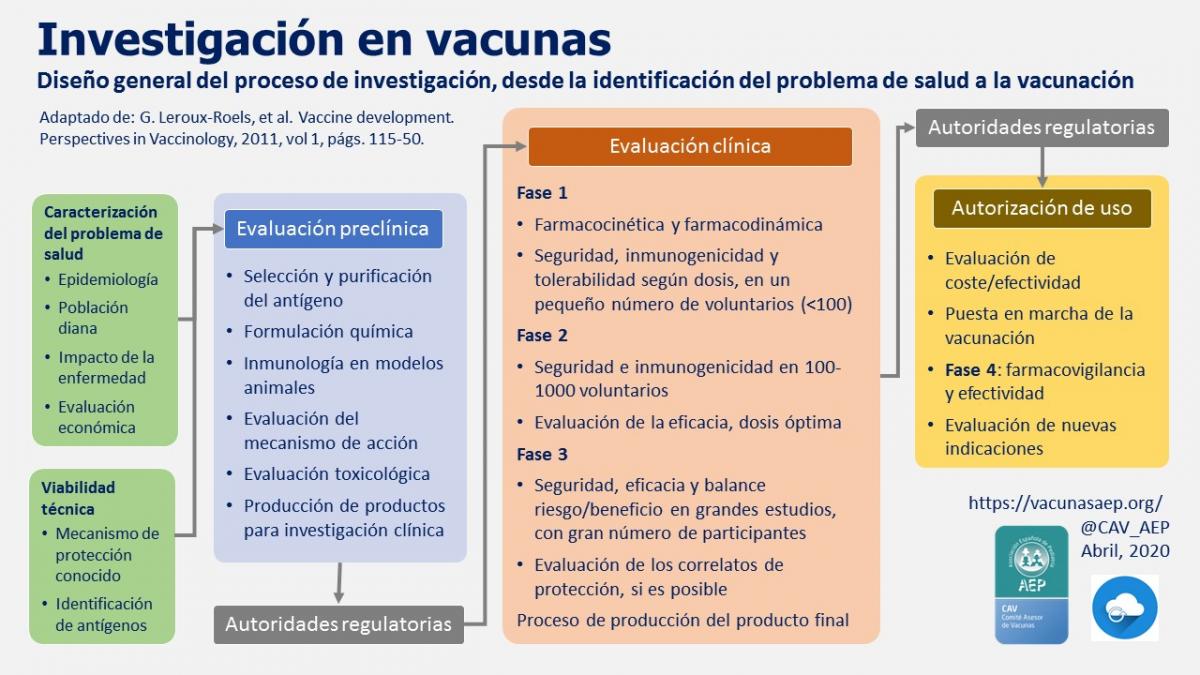
6. Bibliografía
- Agencia Española de Medicamentos y Productos Sanitarios. Buena práctica clínica.
- Agencia Española de Medicamentos y Productos Sanitarios. Normas de Buena Práctica Clínica (CPMP/ICH/135/95).
- Beauchamp TL, et al. Principles of Biomedical Ethics. Oxford University Press; 2019.
- Bonhoeffera J, et al. Brighton Collaboration Clinical Trial Protocol Working Group. Template protocol for clinical trials investigating vaccines—Focus on safety elements. Vaccine. 2013;31:5602-20.
- Consejo Superior de Investigaciones Científicas (CSIC). Ética en la investigación.
- Council for International Organizations of Medical Sciences (CIOMS). International Ethical Guidelines for Health-related Research Involving Humans. Geneva: CIOMS; 2016.
- Dirección General de Salud Pública, Calidad e Innovación y Agencia Española de Seguridad Alimentaria y Nutrición. Ministerio De Sanidad, Consumo y Bienestar Social. Dirección General de Biodiversidad y Calidad Ambiental. Ministerio para la Transición Ecológica. Programa de Cumplimiento de las Buenas Prácticas de Laboratorio Noviembre 2018 (Ampliación del alcance del programa de cumplimiento de BPL).
- Egger GF, et al. European Union Clinical Trials Register: on the way to more transparency of clinical trial data. Expert Rev Clin Pharmacol. 2013;6:457-9.
- Encouraging Vaccine Innovation, § 3093 of the 21st Century Cures Act (Dec. 13, 2016).
- Encouraging Vaccine Innovation: Promoting the Development of Vaccines that Minimize the Burden of Infectious Diseases in the 21st Century. Report to Congress. December 2017.
- European Commission. Good Manufacturing Practice (GMP) guidelines "The rules governing medicinal products in the European Union" EudraLex - Volume 4.
- European Medicines Agency. Guideline on clinical evaluation of new vaccines EMEA/CHMP/VWP/164653/05 Rev. 1, 2018.
- Evidence-Based Medicine Working Group. Evidence-based medicine. A new approach to teaching the practice of medicine JAMA. 1992;268:2420-5.
- Giaquinto C, et al. Registration of Vaccines, Safety Follow-Up, and Pediatric Investigation. Plan. En: T. Vesikari, P. Van Damme (eds.), Pediatric Vaccines and Vaccinations. Springer International Publishing AG 2017. p 251-7.
- Goetz K, et al. First-in-human clinical trials with vaccines—what regulators want. Nat Biotechnol. 2010;28: 910-6.
- Guyatt GH, et al. GRADE Working Group. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. Br Med J. 2008;336:924-6.
- Harrington L, et al. Recruitment barriers for prophylactic vaccine trials: A study in Belgium. Vaccine. 2017;35(48 Pt B):6598-603.
- James M, et al. Vaccine Production: Main Steps and Considerations. En: Bloom B and Lambert P. The Vaccine Book. Chapter 5. 2.ª ed. 2016 Elsevier Inc. p 77-96.
- Karlberg J , et al. Revisión de ensayos clínicos: una guía para el Comité de Ética. Hong Kong, RP de China, ed Karlberg, Johan Petter Einar, 2010.
- Lazcano-Ponce E, et al. Ensayos clínicos aleatorizados: métodos de aleatorización, análisis, variantes, consideraciones éticas y regulación Salud Publica Mex. 2004;46:559-84.
- Ministerio de Sanidad. Agencia Española de Medicamentos y Productos Sanitarios. Documento de instrucciones de la Agencia Española de Medicamentos y Productos Sanitarios para la realización de ensayos clínicos en España. Versión 16, de 31 de enero de 2022.
- Mitchell V, et al. The Children's Vaccine Initiative: Achieving the Vision. Committee on the Children's Vaccine Initiative: Planning Alternative Strategies, Institute of Medicine. Sanford, Editors; Washington, D.C. 1993.
- Molina Arias M, et al. Ensayo clínico (I). Definición. Tipos. Estudios cuasiexperimentales. Evid Pediatr. 2014;10:52.
- Myron M Levine, et al. How are Vaccines Assessed in Clinical Trials?. En: Bloom B and Lambert P. The Vaccine Book. Chapter 6. 2.ª ed. 2016 Elsevier Inc. p 97-119.
- Real Decreto 1090/2015, de 4 de diciembre, por el que se regulan los ensayos clínicos con medicamentos, los Comités de Ética de la Investigación con medicamentos y el Registro Español de Estudios Clínicos.
- Reglamento (UE) N.º 536/2014 del Parlamento Europeo y del Consejo de 16 de abril de 2014 sobre los ensayos clínicos de medicamentos de uso humano. Diario Oficial de la Unión Europea L 158/1- L 158/74.
- Rolling K, et al. The vaccine development process. J Am Pharm Assoc (2003). 2016;56:687-9.
- Sackett DL, et al. Evidence-based medicine: how to practice and teach EBM. Churchill Livingstone, 2000.
- Shimabukuro TT, et al. Safety monitoring in the Vaccine Adverse Event Reporting System (VAERS). Vaccine. 2015;33:4398–405.
- Singh K , et al. The clinical development process for a novel preventive vaccine: An overview. J Postgrad Med. 2016;62:4–11.
- Straus SE, et al. Evidence-Based Medicine: How to Practice and Teach It. Churchill Livingstone, 2010. p 1.
- World Health Organization. Expert Committee on Biological Standardization. Sixty-seventh report. Annex 1. WHO guidelines on clinical evaluation of vaccines: regulatory expectations. WHO Technical Report Series, N º 924, 2004.
- World Health Organization. Expert Committee on Biological Standardization. Sixty-seventh report. Annex 9. Guidelines on clinical evaluation of vaccines: regulatory expectations. WHO Technical Report Series, N.º 1004, 2017.
- World Health Organization. Expert Committee on Biological Standardization. Sixty-seventh report. Annex 10. Human challenge trials for vaccine development: regulatory considerations. WHO Technical Report Series, N.º 1004, 2017.
- World Health Organization. WHO’s vision and mission in immunization and vaccines 2015-2030.
- World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191-4.
7. Enlaces de interés
-
Autoridades regulatorias de fármacos:
8. Historial de actualizaciones
| 20 de abril de 2020 | Creación de todos los apartados del capítulo |
| 25 de enero de 2023 | Cambio de nombre a Manual de Inmunizaciones |
| 24 de agosto de 2023 | Revisión de todos los apartados y creación de un nuevo apartado "Consentimiento y asentimiento informado en ECA de vacunas". Nuevas citas bibliográficas |
-oOo-

